| GRJ top > 遺伝子疾患情報リスト | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
シトリン欠損症
|
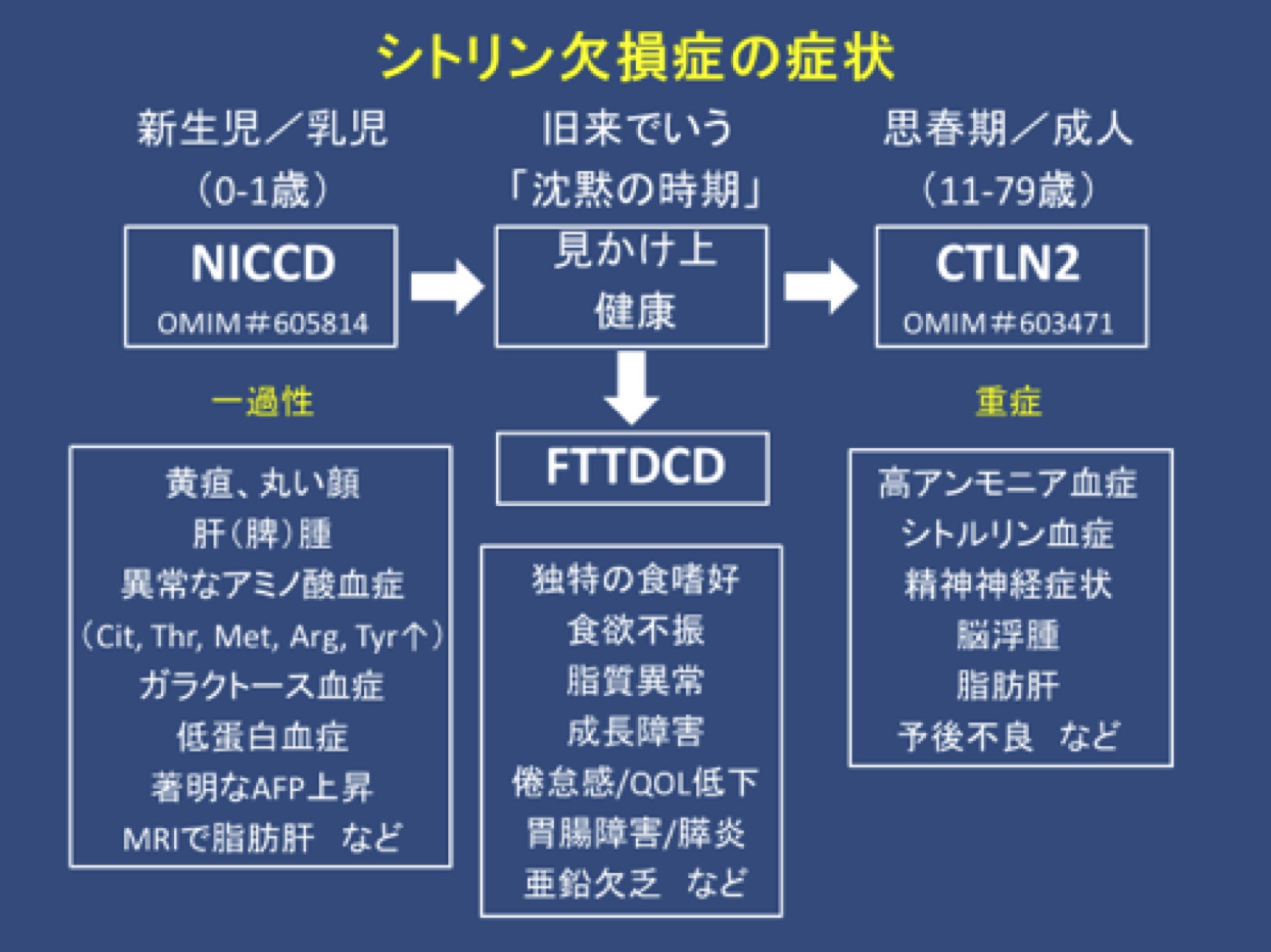
| 臨床型 (年齢) |
血中もしくは血漿アンモニア濃度(μmol/L) | 血漿もしくは血清シトルリン濃度(μmol/L)1 | 血漿もしくは血清アルギニン濃度(μmol/L) | 血漿もしくは血清スレオニン/セリン比 | 血清膵分泌性トリプシンインヒビター(PSTI)濃度2(ng/mL) |
| 対照群 | 18-473 | 17-433 | 54-1303 | 1.10 | 4.6-20 3 |
| NICCD(0-6ヶ月) | 60 | 300 | 205 | 2.29 | 30 |
| FTTDCD(1-11歳) | 正常もしくは僅かに上昇 | 正常もしくは僅かに上昇 | 通常は正常 | 不明 | 不明 |
| CTLN2(11-79歳) | 152 | 418 | 198 | 2.32 | 71 |
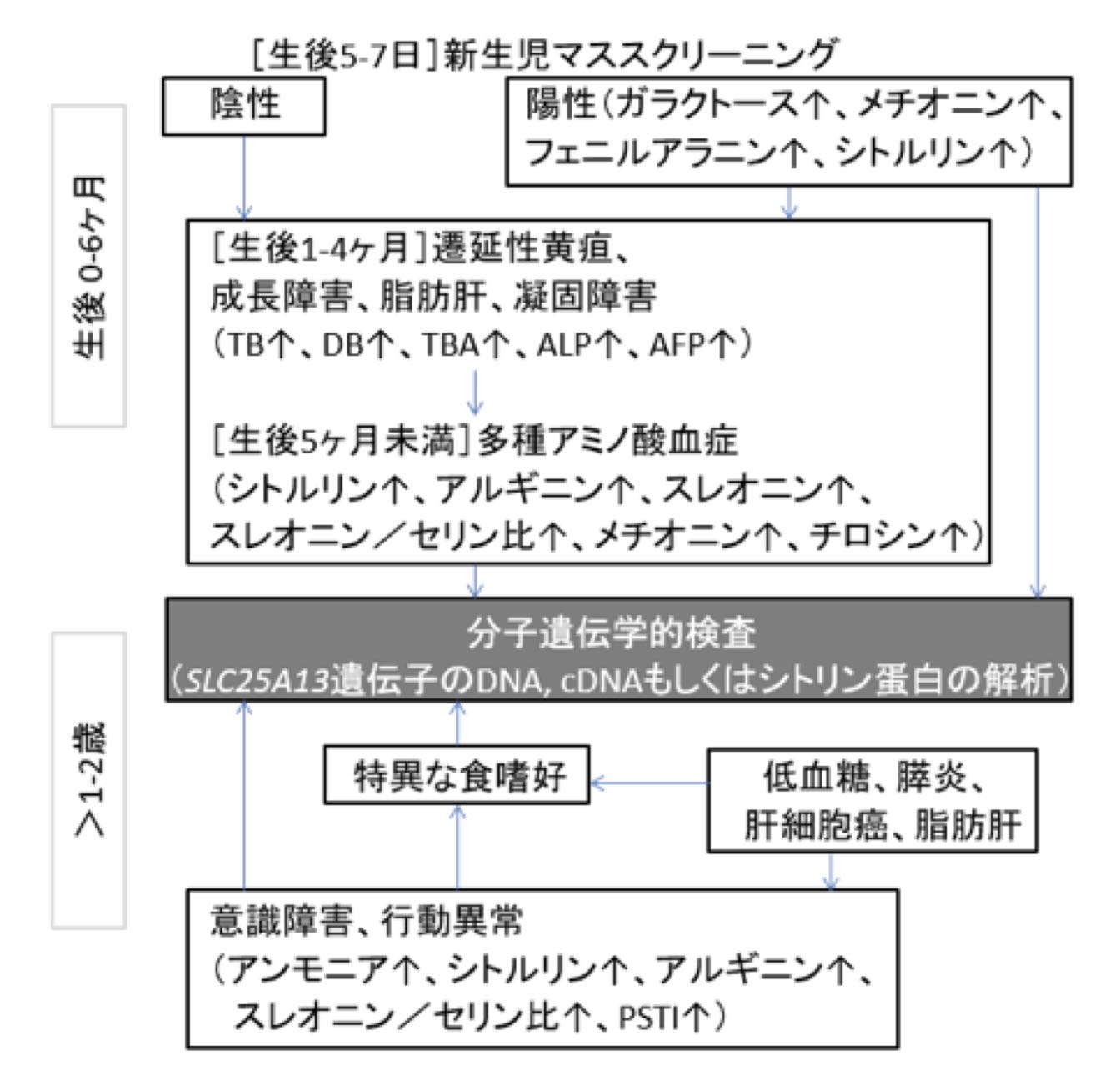
- 新生児スクリーニングで見つけることができるシトルリン血症は、NICCDの最も早く確認できる生化学的異常である。
- NICCD患者の一部やCTLN2発症前の患者では血清PSTI濃度高値であるため、血清PSTI濃度測定はCTLN2の発症前診断に有用である可能性がある。
- 範囲
表2 NICCDの生後0-6ヶ月における血漿スレオニン、メチオニン、チロシン濃度
| アミノ酸 | 中間値(25%-75%範囲)(μmol/L) | 対照群の範囲(μmol/L) |
| スレオニン | 496 (291-741) | 67-190 |
| メチオニン | 124 (53-337) | 19-40 |
| チロシン | 178 (99-275) | 40-90 |
分子遺伝学的検査
分子遺伝学的検査の手法には、単一遺伝子検査や多遺伝子パネルの利用などがある。
単一遺伝子検査 SLC25A13遺伝子のシークエンス解析がまず行われ、変異が見つからないか1つのみの場合はつづいて欠失・重複解析を行う。
日本人もしくは中国人では、最初に病原性変異の標的遺伝子解析を行うことができる。
- 日本人のシトリン欠損症患者では、病原性変異の多く(~70%)を2つの病原性アレルが占める[著者の個人的な経験]。
- 中国人のシトリン欠損症患者では、80%超の病原性変異を4つの病原性アレルが占める。
SLC25A13遺伝子や他の関心領域の遺伝子(「鑑別診断」を参照)を解析できる多遺伝子パネルを考慮することがある。注:(1)パネルに含まれる遺伝子や検査の診断感度は検査施設によって異なり、時間とともに変化する傾向にある。(2)一部の多遺伝子パネルには、 このGeneReviewで触れていない病態と関連する遺伝子も含まれている可能性がある。そのため、臨床医はどの多遺伝子パネルがもっとも合理的なコストでその病態の遺伝的な原因を追究できるか見極める必要があるが、一方で意義不明の変異や基本的な臨床型を説明できない遺伝子変異の同定は制限される。(3)一部の検査施設ではオプションとして、その施設向けにデザインされたパネル、および/もしくは臨床医によって指定された遺伝子を含めて臨床型に的を絞ったエクソーム解析を採用していることがある。(4)パネルに用いられている方法は、シークエンス解析、欠失/ 重複解析、およびその他シークエンシングに基づかない検査である。
シークエンス解析では同定されないだろうこの疾患に関連するSLC25A13遺伝子の大きな欠失/重複の頻度から、欠失/重複解析も含めた多遺伝子パネルが推奨される(表3を参照)。
多遺伝子パネルに関するイントロダクションについてはここをクリック。遺伝子検査をオーダーする臨床医のための詳細な情報についてはここで閲覧することができる。
表3 シトリン欠損症で用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | この方法で同定される変異2>をもつ発端者の割合 |
SLC25A13 |
シークエンス解析3 | 85%-90%4 |
| 標的遺伝子の欠失/重複解析5 | 10%-15%6 |
- 染色体遺伝子座と蛋白については表A「遺伝子およびデータベース」を参照。
- この遺伝子で同定されるアレル変異に関する情報は「分子遺伝学」を参照。
- シークエンス解析では、良性/おそらく良性/意義不明/おそらく病原性をもつ/病原性をもつ変異を同定する。病原性変異には、小さな遺伝子内欠失/重複、ミスセンス、ノンセンス、スプライス部位変異などがある。典型的には、エクソンもしくは全遺伝子の欠失や重複は同定されない。シークエンス解析結果の解釈について考慮すべき問題についてはこちらをクリック。
- Songら(2013)、Linら(2016)
- 標的遺伝子の欠失/重複解析では遺伝子内欠失/重複を同定する。用いられる方法には、定量PCR、ロングレンジPCR、MLPA(multiplex ligation-dependent probe amplification)法、単一エクソンの欠失/重複を同定する標的遺伝子マイクロアレイ(gene-targeted microarray)などがある。
- Takayaら(2005)、Tabataら(2008)、Wongら(2008)、Songら(2013)、Linら(2016)、Zengら(2016)、Zhengら(2016)、Zhangら(2017)
ウェスタンブロット解析
生化学的検査はシトリン欠損症に合致するものの、分子遺伝学的検査でSLC25A13遺伝子変異が1つも同定されない/1つしか同定されない稀な例においては、シトリン蛋白に対するウェスタンブロット解析を考慮することができる。
アミノ末端側に特異的な抗ヒトシトリン抗体を用いたウェスタンブロット解析では、SLC25A13遺伝子両アレル変異を有する患者の肝臓や培養線維芽細胞において交差反応性免疫物質をほとんどもしくは全く検出しない。ウェスタンブロット解析で用いられるその他の試料には末梢血リンパ球がある。シトリン蛋白は、培養リンパ球から抽出したミトコンドリア蛋白を用いたウェスタンブロット解析にて、より容易に検出できる可能性がある。
臨床像
臨床記述
シトリン欠損症は、新生児期/乳児期にシトリン欠損による新生児肝内胆汁うっ滞症(NICCD)を、年長児ではシトリン欠損による成長障害と脂質異常症(FTTDCD)を、成人期では成人発症Ⅱ型シトルリン血症(CTLN2)として精神神経症状を伴う反復性高アンモニア血症を呈する。しばしばFTTDCDとCTLN2では、高蛋白や高脂質の食事を好み、炭水化物を嫌う食嗜好が特徴とされる。CTLN2患者では、NICCD/FTTDCDの既往があることもないこともある。CTLN2に移行するNICCD/FTTDCD患者の比率は不明である。食事管理以外に特別な医学的配慮を必要としないNICCD患者では、生化学的異常が続く間はこまめな経過観察が推奨される。
シトリン欠損症による新生児肝内胆汁うっ滞症(NICCD)
1歳未満のNICCD患児は一過性の肝内胆汁うっ滞を呈する(表4を参照)。その他の所見には、肝腫大を伴うびまん性脂肪肝、肝線維化を合併する肝実質への細胞浸潤、低出生体重、成長遅滞、低蛋白血症、凝固因子の低下、溶血性貧血、様々な重症度の肝機能障害(主に軽症)、低血糖などがある。
表4 生後0-6ヶ月のNICCD患児における肝機能検査
| 検査項目 | 中間値(25%-75%範囲)(mg/dL) | 対照群の範囲(mg/dL) |
| NICCDにおけるTB | 4.9 (2.8-8.0) | 0.2-1.0 |
| NICCDにおけるDB | 2.5 (1.5-3.7) | 0-0.4 |
| NICCDにおけるTB/DB比 | 0.55 (0.41-0.66) | - |
| TBA | 239 (172-293) | 5-25 |
| AFP | 91,900 (33,200-174,700) | 260-6,4001, 2 2-552, 3 |
TB=総ビリルビン
DB=直接ビリルビン
TBA=総胆汁酸
AFP=αフェトプロテイン
- 生後0-1ヶ月
- Tamamoriら(2002)
- 生後2ヶ月以降
NICCDは一般的に重症ではないが、まれに肝移植が必要となる。脂溶性ビタミンの補給や乳糖除去ミルクの使用(続発性高ガラクトース血症の患者)、中鎖脂肪酸(MCT)強化ミルクなどの治療によって、症状は典型的には1歳までに軽快する。
1-2歳頃から、患児は高蛋白・高脂質食を好み、糖分や炭水化物の多い食事を嫌うようになる。
10代以降、シトリン欠損症患者の中には精神神経症状を伴った重症CTLN2に進展する者もいる。典型的には、NICCDに続く適応期(代償期)からCTLN2発症への移行は緩徐であるが、CTLN2は通常突然に発症する。
シトリン欠損症による成長障害と脂質異常症(FTTDCD)
FTTDCDはNICCD発症後およびCTLN2発症前の新しい臨床型として最近提唱された(Songら, 2011年)。FTTDCDの臨床および検査上の特徴はまだ明らかではない。この時期(旧来はCTLN2発症前の「見かけ上健康」な時期とされてきた)の間に、検査や臨床所見で異常を認める患児もいる。
検査では、トリグリセリド高値・総コレステロール高値・LDL高値・HDL低値といった脂質異常や、そのほか乳酸/ピルビン酸比の上昇、尿中酸化ストレスマーカーの高値、TCA回路代謝物の著しい偏りなどが認められる。
臨床的な異常には、成長遅滞、低血糖、膵炎などがある。適応期・代償期(旧来「沈黙」の時期と見なされていた)のシトリン欠損症患児において、重度の倦怠感やQOLの低下を認めることが明らかとなった。さらに、制限型の神経性食欲不振症に類似した重篤な食欲不振と体重減少で発症した12歳女児例が報告されている。
成人発症Ⅱ型シトルリン血症(CTLN2)
CTLN2は、反復性の高アンモニア血症や、夜間せん妄、異常行動(攻撃性・易刺激性・過活動)、妄想、見当識障害、不穏、眠気、記憶喪失、羽ばたき振戦、けいれん発作、昏睡など、肝性脳症や遺伝性尿素サイクル異常と極めて類似した精神神経症状を呈する。脳CTは正常で、脳波ではびまん性の徐波を認める。
発症は突発的で、通常は20-50歳時に起こる(範囲:11-79歳、平均年齢34.4±12.8歳、n=103)。
CTLN2患者の多くは、高蛋白・高脂質食(豆・ピーナッツ・卵・ミルク・チーズ・魚・肉など)を好み、米・ジュース・甘い物など高炭水化物食を嫌うという強い食嗜好がある。症状はしばしばアルコール、糖類、薬物の摂取や外科手術によって誘発される。
ほとんどの患者はやせている。90%を超える患者ではBMIは20未満で、約40%は17未満である(範囲:15.6-19.1, n=110)(健康な日本人では男性20-24、女性19-23)。
10%を超えるCTLN2患者では以下の合併症を認める。
- 膵炎 CTLN2発症前に若年発症慢性膵炎と肝硬変を伴わない肝細胞癌を認めることがある。
- 高脂血症 高炭水化物を摂取したシトリン欠損症患者で高トリグリセリド血症はしばしば認められる。
- 脂肪肝 NICCDおよびCTLN2患者のほとんどで、組織学的にNASH(非アルコール性脂肪性肝炎)に合致する脂肪肝を認める。軽度の線維化もまた認めうる。
- 肝細胞癌 CTLN2と診断される前であっても肝細胞癌を認めることがある。
肝内胆汁うっ滞はまれである。しかし、なかには後から振り返ってみると幼少期にNICCDの徴候を認めていた者もいる。例えば、肝移植を施行した16歳のCTLN2患者において、乳児期早期に一過性の低蛋白血症および黄疸を認めていた。
検査異常所見
- 肝臓の膵分泌性トリプシンインヒビター(PSTI)濃度は上昇している(表1を参照)。
注:CTLN2患者の肝臓では、PSTI mRNAは30-140倍増加している。 - 分岐アミノ酸の減少により、血漿/血清フィッシャー比(分岐鎖アミノ酸[BCAAs]:バリン+ロイシン+イソロイシン、芳香族アミノ酸[AAAs]:トリプトファン+フェニルアラニン)は~3.4から~2まで低下している。
- 肝特異的アルギニノコハク酸合成酵素(ASS)活性は対照群の約10%にまで低下している(SLC25A13遺伝子変異の二次的な効果)。
- 血漿αフェトプロテイン濃度は、肝細胞癌を合併している場合を除き、ほとんどすべてのCTLN2患者において正常である。
病理所見では、肝機能障害をほとんど認めないか全く認めないにもかかわらず、脂肪浸潤や軽度の肝線維化を認める。
臨床記述遺伝子型と臨床型の相関
この疾患では遺伝子型と臨床型との相関は認められていない。
浸透率
患者の性別に関連したCTLN2の臨床型の浸透率には違いがあるようである。
- NICCDの男女比はおおよそ1:1であるが(73:80)、CTLN2の男女比は2.4:1である(120:50)。
- CTLN2の男女比が1:1でないことは、理由は不明だがSLC25A13遺伝子両アレル変異を有する患者では女性のほうが男性よりもCTLN2の臨床型を呈しにくいことを示唆している。
命名法
NICCD NICCDは、分子遺伝学的検査でSLC25A13遺伝子両アレル変異の存在が確認される前は「原因不明の脂肪肝を伴う特発性新生児肝炎」[Ohuraら1997]として知られていた。
CTLN2 Miyakoshiら[1968]は、高アンモニア血症と独特の慢性反復性肝脳変性症を認める患者において血中シトルリン濃度が上昇していることを報告した。この肝脳変性症は、脳の病理学的変化に基づいて「類瘢痕型肝脳疾患」、高度にバランスを欠いた食事に由来する代謝障害もしくは内分泌異常による発達障害に基づいて「栄養障害型肝脳疾患」として知られるようになった。
Sahekiら[1981]は、アルギニノコハク酸合成酵素(ASS)活性/蛋白の定性的および肝特異的な疾患を伴う高シトルリン血症の病型を報告し、のちに「成人発症Ⅱ型シトルリン血症」と命名した。
頻度
日本におけるSLC25A13遺伝子のホモ変異または複合ヘテロ変異の頻度は、保因者(ヘテロ変異保有者)が65人に1人であることから、17000人に1人と計算される。これはNICCDの頻度と同等であるが、CTLN2の頻度(100,000-230,000人に1人)とは異なる。これらのことから、SLC25A13遺伝子両アレル変異を受け継いだほとんどの日本人はNICCDを発症すると著者らは信じている。 最近まで、シトリン欠損症は日本に限定的であると思われていた。現在ではシトリン欠損症はさまざまな民族に認められることが知られている。新規のSLCA25A13遺伝子変異を有する患者が、イスラエル、パキスタン、米国、英国、中国、チェコ共和国で見つかっている。
中国(65人に1人)、特に中国南部や台湾(48人に1人)、韓国(112人に1人)でも保因者の頻度は高い。
遺伝的レベルでの関連疾患
CTLN2, NICCD, FTTDCDは、現時点でSLC25A13遺伝子変異と関連することが知られる唯一の臨床型である。
鑑別診断
シトリン欠損症でみられる血漿シトルリン濃度の上昇は以下でも認められる。
- シトルリン血症Ⅰ型(CTLN1、ASS欠損症) CTLN1はオーバーラップする幅広い臨床型を呈する。急性新生児型(「古典」型)、軽症遅発型(非「古典」型)、症状や高アンモニア血症のない病型、妊娠中/分娩後の女性に発症する重症型がある。
- 新生児(”古典”)型 出生後すぐに、急性新生児型の患児は高アンモニア血症およびその合併症を呈し、迅速な治療が行われなければ死亡する。迅速に治療が行われれば期間は不明確だが生存することがある。しかし、通常は著しい神経学的後遺症を残す。
- 非古典型 遅発型では、高アンモニア血症のエピソードは急性新生児型でみられるのと同様であるが、初期の神経所見はよりわずかである可能性がある。
CTLN1は、尿素サイクルの第3段階でシトルリンとアスパラギン酸からアルギニノコハク酸を合成するASSが欠損する。未治療のCTLN1患者は、高アンモニア血症、血漿シトルリン濃度上昇、血漿アルギニン濃度低下を呈する。CTLN1はASS1遺伝子の両アレル変異で発症する常染色体劣性遺伝性疾患である。(注:CTLN2では、機序は不明だがASS蛋白が肝特異的に欠損する。肝ASS mRNAまたはASS1遺伝子には異常は存在しない)
この病態は、血漿アルギニン低値を含めた生化学的検査によってシトリン欠損症と鑑別することができる。
- 新生児(”古典”)型 出生後すぐに、急性新生児型の患児は高アンモニア血症およびその合併症を呈し、迅速な治療が行われなければ死亡する。迅速に治療が行われれば期間は不明確だが生存することがある。しかし、通常は著しい神経学的後遺症を残す。
- アルギニノコハク酸尿症(アルギニノコハク酸リアーゼ[ASL]欠損症)(尿素サイクル異常総論を参照)
- リジン尿性蛋白不耐症(LPI)
- ピルビン酸カルボキシラーゼ(PC)欠損症
- 腎不全
- 古典的ガラクトース血症 シトリン欠損症で発症した古典的ガラクトース欠損症の新生児が報告されている。
シトリン欠損症でも尿素サイクル異常と同様に、蛋白質やその他の窒素含有分子の崩壊によって生成された窒素の代謝異常から高アンモニア血症を呈する(尿素サイクル異常総論を参照)。尿素サイクルの初め4つの酵素(CPSI, OTC, ASS, ASL)のいずれか、オルニチントランスポーター、もしくは補因子生成酵素(NAGS)の重度な欠損もしくは完全な欠損により、ほとんどの患者において生後数日以内にアンモニアやその他の代謝前駆体の蓄積が生じる。
シトリン欠損症で見られる新生児/乳児胆汁うっ滞は以下の疾患でも認められる。
- 特発性新生児肝炎(INH)および肝外胆道閉鎖症(EBA) INHやEBAと比較してNICCDでは血清直接ビリルビンやALTは低く、血清総胆汁酸やALPは高い。NICCDはまた、INHより血清γ-GTPは高く血清ASTは低い。
- 進行性家族性肝内胆汁うっ滞症(PFIC) NICCDでは血清γ-GTPは高値で、PFICや良性反復性肝内胆汁うっ滞症(BRIC)を含むγ-GTP低値~正常の他の肝内胆汁うっ滞症と鑑別される。PFICはATP8B1遺伝子の両アレル変異によって起こる(ATP8B1欠損症を参照)。PFIC2はABCB11遺伝子の両アレル変異によって起こる(OMIM 601847を参照)。BRICの一部はATP8B1遺伝子の両アレル変異で起こる場合がある。
体質性黄疸および高ビリルビン血症はビリルビンの代謝異常で起こる。非抱合型(間接型)優位の高ビリルビン血症(UDP-グルクロン酸転移酵素1欠損)と抱合型(直接型)優位の高ビリルビン血症(毛細胆管膜のATP依存性トランスポーター:ABCC2[OMIM 601107], ABCB11[OMIM 603201], ATP8B1[ATP8B1欠損症を参照]の欠損)がある。
その他
- 血管造影により門脈体循環短絡は除外することができる。これらの短絡路とは、門脈から下大静脈/肝静脈/脾静脈への異常な血流のことを指す。シトリン欠損症患者では画像検査(超音波検査、MRIなど)でそのような短絡路は認められない。
- 30%を超えるCTLN2患者は初めてんかん発作や精神疾患(うつ病、統合失調症など)と誤診されてきた。その他、肝細胞癌、膵炎、高脂血症と診断されている可能性がある。
臨床的マネジメント
初期診断に続く評価
シトリン欠損症の臨床型であると診断された患者において、疾患の広がりやニーズを把握するため、以下のような評価が推奨される。
NICCD
- 肝臓、脾臓のサイズを評価する。
- 腹部エコー、CT、MRIによって脂肪肝を評価する。
- 食事内容を評価する。
FTTDCD
- 詳細な身体計測と年齢・性別の成長曲線を用いた評価。
- 食事内容を評価する。
CTLN2 食事の炭水化物、蛋白質、脂質の比率を評価する。
全員 臨床遺伝専門医および/または遺伝カウンセラーへの診療依頼。病変に対する治療
NICCD ほとんどのNICCD患児では、脂溶性ビタミンの補充や乳糖除去ミルク、MCT強化ミルクによって症状は生後12か月までに軽快する。
母乳からプロリンを豊富に含むミルクへの変更で軽快した同胞2人の症例が報告されている。
NICCD患児のなかには無治療で軽快する者もいる。母乳や一般ミルクを減らし、同時に卵や肉のような高蛋白・高脂質の固形物の摂取を開始したことがシトリン欠損症患者に有益だった可能性がある。
治療用ミルクは生涯にわたって必要とするわけではない。ほとんどのNICCD患児は、蛋白・脂質の豊富な加工食/固形食が開始されている1歳までには臨床的かつ生化学的に軽快する。1歳をすぎても治療を行うことでFTTDCDやCTLN2発症の可能性を減らせるかどうかは現時点で不明である。
さらに、NICCDでは亜鉛欠乏もよく認められるため、血液検査で亜鉛欠乏が示唆される場合、とくに著明な成長障害を認める場合には亜鉛の補給を推奨するべきである。
NICCDおよび重度肝機能障害を認める乳児4人において、原因不明のチロシン血症と診断され生後10-12ヶ月に肝移植を施行されたことが報告されている。
FTTDCD この新しいシトリン欠損症の臨床型に対する治療法の記述はほとんどない。
・FTTDCDの幼児は固有の食嗜好がある(米を嫌い魚を好むなど)。成長障害は次第に改善し、3歳時には3パーセンタイルを超える。脂質異常もまた徐々に軽快する。
・食事療法に加えて、ピルビン酸ナトリウムの投与は成長遅滞の改善に効果的である可能性がある。ピルビン酸ナトリウムは肝細胞におけるNADH/NAD+比を低下させるが、これはシトリン蛋白の生成に極めて重要な変化であり、成長遅滞に対して改善効果を示す可能性がある。
CTLN2 現在までにもっとも成功した治療法は肝移植である。肝移植により高アンモニア血症クリーゼは予防され、代謝異常は改善し高蛋白食を好む食嗜好はなくなる。過去にはおよそ全てのCTLN2患者は肝移植を必要としたが、アルギニンやピルビン酸ナトリウムの導入やMCTオイルの投与で状況は変わった。そのほかの治療は以下が挙げられる。
- アルギニンの投与(5-10g/日)は血中アンモニア濃度を下げるのに有効であると報告されている。
- カロリー/炭水化物摂取を減らし蛋白摂取を増やすと高トリグリセリド血症は改善する。
- ピルビン酸ナトリウムの投与(4-9g/日)は高アンモニア血症の頻度を減らし、成長の改善に有効であることがいくつかの症例で報告されている。
- MCTオイル(85%MCTを含むマクトンオイル45mL/日)の投与により、検査所見がすべて正常化する完全回復、もしくは高アンモニア血症の症状がない改善が認められる。
一次病変の予防
高アンモニア血症を予防し成長障害を改善させるためには、高蛋白・高脂質食および低炭水化物食が推奨される。
高炭水化物食およびアルコールは避けるべきである。
高アンモニア血症クリーゼの予防には、アルギニンの投与が効果的である可能性がある。
二次合併症の予防
ビタミンD欠乏および亜鉛欠乏はNICCDでよくみられる合併症である。重症感染および肝硬変もまた一部のNICCD患者の致死的合併症として報告がある。それゆえ、ビタミンDおよび亜鉛の補給、急性感染症の制御がNICCD患者では推奨される。
経過観察
1歳以上のシトリン欠損症患者において、FTTDCDの病像を認めないか経過観察する。こまめに身体計測/検査(身長、体重、頭囲、トリグリセリド・総コレステロール・HDLコレステロール・LDLコレステロールを含む血清脂質濃度など)を行うことがのぞましい。
数ヶ月ごとに以下を測定することが推奨される。
- 血漿アンモニア濃度(とくに夕方もしくは食後2時間)
- 血漿シトルリン濃度
- 血清PSTI濃度
血漿シトルリン濃度や血清PSTIの上昇はCTLN2発症を示唆し、迅速に治療を開始するべきである。
回避すべき薬物や環境
低蛋白/高カロリー(高炭水化物)食 低蛋白/高カロリー食は、尿素サイクル酵素欠損による高アンモニア血症の予防の一助となるが、全てのシトリン欠損症患者(すなわちNICCD, FTTDCD, CTLN2)には有害である。高炭水化物食はNADH産生を増加させ、尿素生成を阻害し、リンゴ酸-クエン酸シャトルを促進し、高アンモニア血症、脂肪肝、高トリグリセリド血症をきたす可能性がある。
グリセロール、フルクトース、グルコースのような糖類の静注 グリセロールを含んだ高浸透圧剤を重症脳浮腫に使用すると悪化してしまうため、CTLN2患者では禁忌である。大量のグリセロールやフルクトースが分解されると肝の細胞質でNADHが産生され、これにより肝機能は抑制される可能性がある。
高濃度グルコースの静注もまた高アンモニア血症を悪化させることがある。
注:マンニトール静注はより安全なようである。
アルコール アルコール脱水素酵素(ADH)は肝の細胞質でNADHを産生するため、アルコール摂取はCTLN2発症の誘因となりうる。
薬物 アセトアミノフェンやラベプロゾールはCTLN2の誘因となるかもしれない。
リスクのある血縁者の評価
シトリン欠損症の症候はないが疾患リスクのある同胞では、発症前に乳児期からの適切な食事管理(母乳栄養の中止および乳糖除去ミルク・MCT強化ミルクの開始)を行うことができるよう、遺伝学的状況を明らかにすることがのぞましい。
シトリン欠損症の無症候/発症前の患者ではたいてい生化学的異常を認めることはないので、疾患リスクのある血縁者(同胞など)の確定診断には、発端者のSLC25A13遺伝子の分子遺伝学的所見に重きが置かれるだろう。
- もし発端者のSLC25A13遺伝子変異が両方とも同定されているのならば、分子遺伝学的検査は信頼性をもって行うことができる。
- もし発端者でSLC25A13遺伝子変異が1つしか確認されていないのならば、疾患リスクのある血縁者が1つの遺伝子変異を有する場合にシトリン欠損症を除外することは適切ではない可能性がある。そのようなケースでは、確定診断もしくは診断の除外のために、臨床的および生化学的な評価を経時的に行っていくことが必要であろう。
- もし発端者でSLC25A13遺伝子変異が確認されていない場合には、疾患リスクのある血縁者に対する分子遺伝学的検査は有用ではないだろう。そのようなケースでは、確定診断もしくは診断の除外のために、臨床的および生化学的な評価を経時的に行っていくことが必要であろう。
遺伝カウンセリングとして扱われるリスクのある血縁者への検査に関する問題は「遺伝カウンセリング」の項を参照のこと。
研究中の治療法
疾患や病態の広範囲にわたる臨床試験に関する情報は、米国ではClinicalTrials.govを、欧州ではwww.ClinicalTrialsRegister.euを参照のこと。注:この疾患に対する臨床試験は行われていない可能性がある。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
シトリン欠損症は常染色体劣性遺伝形式で遺伝する。
患者家族のリスク
発端者の両親
- 罹患者の両親は必然的にヘテロ接合体保有者である(すなわち1つのSLC25A13遺伝子変異をもつ保因者)。
- まれに親はCTLN2の重篤な症状がなく2つのSLC25A13遺伝子変異を有していることがある。NICCDの日本人家族163人において、父親48人のうち2人、母親54人のうち1人で見つかっている。無症候の父親はNICCD患者である息子と同じSLC25A13遺伝子変異を有していることが判明した。
- ヘテロ接合体保有者(保因者)は無症状である。
発端者の同胞
- 両親ともSLC25A13遺伝子変異の保因者であった場合、受胎時に、発端者の同胞が2つの病原性変異を受け継いでいる確率は25%、無症候性保因者である確率は50%、25%は非罹患者および非保因者である確率は25%である。
- 片親が保因者で片親が2つのSLC25A13遺伝子変異を有している場合、受胎時に、発端者の同胞が2つのSLC25A13遺伝子変異を有する確率は50%、1つの変異を有する無症候性保因者である確率は50%である。
- 概して、SLC25A13遺伝子の両アレル変異を受け継いだ同胞は、発端者と同様にシトリン欠損症を発症し臨床症状を呈するようになる。しかし、浸透率が低いことや家族内で臨床的な重症度に幅があることが報告されている。
- ヘテロ接合体保有者(保因者)は無症状である。
発端者の子
もしシトリン欠損症患者で罹患者もしくは保因者である子どもがまだいないのならば、生まれてくる子どもは必然的にSLC25A13遺伝子変異のヘテロ接合体保有者(保因者)であるだろう。
発端者の他の家族
発端者の両親の同胞がSLC25A13遺伝子変異の保因者であるリスクは50%である。
保因者(ヘテロ接合体保有者)診断
疾患リスクのある血縁者に保因者診断を行うには、事前に家族内のSLC25A13遺伝子変異が同定されている必要がある。
遺伝カウンセリングに関連した問題
早期診断・治療目的の疾患リスクのある血縁者に対する検査についての情報は「臨床的マネジメント」「リスクのある血縁者の評価」を参照のこと。
家族計画
- 遺伝学的リスクの評価や遺伝学的状況の明確化、および出生前診断の有用性についての議論を行う最適な時期は妊娠前である。
- 罹患者、保因者、保因者のリスクがある若年成人に対して、遺伝カウンセリング(潜在的な子どもへのリスクや出産方法の選択肢に関する話し合いを含む)を申し出ることがのぞましい。
DNAバンキング
DNAバンクは(主に白血球から調整した)DNAを将来利用することを想定して保存しておくものである。検査技術や遺伝子、アレル変異、および疾患に対するわれわれの理解が将来さらに進歩すると考えられるので、罹患者のDNA保存を考慮すべきである。
出生前診断および着床前診断
ひとたび家族内でSLC25A13遺伝子変異が同定された場合、リスク妊娠の出生前検査や着床診断を行うことができる。特に早期診断ではなく妊娠中絶を考慮した検査である場合に、医療従事者や家族の間でも出生前検査に関して視点の違いが存在する可能性がある。ほとんどの施設は出生前診断に関する決定は両親の選択によると考えるだろうが、これらの問題に関して話し合うことがのぞましい。
更新履歴
- Gene Review著者:Keiko Kobayashi, PhD, Takeyori Saheki, MD, PhD, and Yuan-Zong Song, MD, PhD
日本語訳者: 和田宏来 (県西総合病院小児科/筑波大学大学院小児科)
Gene Review 最終更新日: 2014.7.31 日本語訳最終更新日: 2016.12.8(minor revision 2017.2.7) - Gene Reviews著者: Takeyori Saheki, MD, PhD, and Yuan-Zong Song, MD, PhD
日本語訳者: 和田宏来 (国際親善総合病院小児科/しんぜんクリニック小児科)
Gene Reviews 最終更新日:2017.8.10. 日本語訳最終更新日: 2020.5.12 (in present)