遺伝性運動失調症総論
(Hereditary Ataxia Overview)
Gene Review著者: Thomas D Bird, MD
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: 2012.11.1. 日本語訳最終更新日: 2013.5.3.
要約
疾患の特徴遺伝性運動失調症は緩徐進行性の失調性歩行を特徴とする遺伝性疾患群であり,しばしば手,会話,眼球運動の協調運動障害を伴う.小脳の萎縮を生じることが多い.本稿では遺伝性運動失調症を,遺伝形式と,原因遺伝子,もしくは染色体座によって分類する.
診断・検査遺伝性運動失調症は,後天性(非遺伝性)運動失調を呈する多くの原因と鑑別しなければならない.遺伝性運動失調症の診断は,家族歴,身体所見,神経画像診断,分子遺伝学的検査によって行う.
臨床的マネジメント
症状の治療: 失調性歩行には杖,歩行器,および車椅子を使用する.筆記,ボタンかけ,食器の使用に際しては,特殊な支援機器が使用できる.構語障害や重度の会話障害がある場合には,言語療法やコンピュータ補助機器を用いる.
一次病変の予防: 遺伝性運動失調症に特異的な治療法は,ビタミンE欠乏を伴う運動失調(AVED)でのビタミンEの投与を除き,存在しない.遺伝カウンセリング
遺伝性運動失調症の遺伝形式は,常染色体優性,常染色体劣性,もしくはX連鎖性である.遺伝カウンセリングとリスクの評価は,患者ごとに確定された遺伝性運動失調症の原因によって異なる.
定義
遺伝性運動失調症の臨床症状
遺伝性運動失調症の臨床症状は,協調運動障害と,開脚歩行を呈する協調運動を欠いた不安定歩行である.四肢と会話に協調運動障害が生じることが多い.
遺伝性運動失調症は,以下の症状が1つ,もしくは複数生じることから生じる.
- 小脳と小脳系器官の機能障害
- 脊髄病変
- 末梢神経性の感覚消失
遺伝性運動失調症の確定診断
遺伝性運動失調症の確定診断には,以下が必要である.
- 神経学的検査で典型的な臨床症状や徴候が確認されること.典型的な臨床症状や徴候には失調性歩行や,指/手の協調運動障害などが含まれ,構語障害や眼振を伴うことが多い.
- 非遺伝性の運動失調の原因が除外されること(「鑑別診断」を参照).
- 運動失調の家族歴,運動失調の原因遺伝子変異の同定,あるいは遺伝性運動失調症に特徴的な臨床像により,疾患が遺伝性であることが判明すること.
注:運動失調の家族歴がない場合,実施可能な遺伝学的検査のすべてに異常を認めないときには,遺伝性であると判断できない.
遺伝性運動失調症の鑑別診断
遺伝性運動失調症の鑑別診断では,運動失調をきたす後天性の非遺伝的原因なども考慮される.このなかにはアルコール依存症,ビタミン欠乏,多発性硬化症,血管障害,原発性あるいは転移性腫瘍,もしくは潜在性の卵巣癌,乳癌,肺癌に伴う腫瘍随伴症候群などがある.
運動失調の後天性の原因は治療できることがあるため,運動失調を呈する患者においてはその可能性を考えてみることが必要である.
遺伝性運動失調症の頻度
オランダにおける常染色体優性の遺伝性小脳性運動失調(ADCA)の頻度は,10万人に対して少なくとも3人と推定されている[van de Warrenburg et al 2002].
原因
遺伝性運動失調症は遺伝形式(常染色体優性,常染色体劣性,X連鎖性,ミトコンドリア遺伝)と病原性変異が生じる遺伝子,あるいは染色体座によって分類される.
Duenas et al [2006],Finsterer [2009a],Paulson [2009],やDurr [2010]も遺伝性運動失調症についてまとめている.
常染色体優性の小脳性運動失調(遺伝性小脳性運動失調[ADCA])
分子遺伝学的な病因が明らかにされる前には,ADCA(遺伝性小脳性運動失調)に対して,Marie失調症,遺伝性オリーブ橋小脳萎縮,小脳-オリーブ萎縮,もしくはより総括的な名称として脊髄小脳変性症などの呼称が使用されていた.
常染色体優性遺伝性小脳失調(ADCA)の分子遺伝学
分子遺伝学的な知見が集積されている常染色体優性の小脳性運動失調を表1にまとめた.ほとんどが脊髄小脳失調症(spinocerebellar ataxias, SCA)であるが,複雑な病型であるDRPLA(歯状核赤核淡蒼球ルイ体萎縮症)や,2つの発作性運動失調症,1つの痙性失調症も含まれている.表1:常染色体優性小脳失調の分子遺伝学
- 遺伝子が不明な場合にのみ,染色体座を掲載.
- 16q22に連鎖を認める複数の日本人家系には,PLEKHG4遺伝子の5’非翻訳領域の一塩基置換(-16C>T)があり,しばしば同じハプロタイプを共有している[Ishikawa et al 2005, Ohata et al 2006].この一塩基置換自体に病的な意義があるのかはまだ不明である[Sakai et al 2010].Edener et al [2011]はSCA4とSCA31が別々の変異から生じていることを示した.これらの変異は同一座(16q22)にある異なった遺伝子に存在している可能性がある.
- 表にはSCA9が入っているが,このタイプのSCAに関する臨床的情報や遺伝学的情報は公表されていない.
- EA2,SCA6,家族性片麻痺性片頭痛の1つのタイプは,すべてCACNA1A遺伝子変異から生じる.
- EA3(眼球の滑動性追従障害を伴う周期性前庭小脳性運動失調)を有する単一家系[Jen et al 2007].
- EA4(回転性めまいと耳鳴を伴う発作性運動失調症)を有する単一家系[Jen et al 2007].
他の常染色体優性の小脳性運動失調(表1以外)
- 小脳性運動失調,難聴,ナルコレプシー,および視神経萎縮を呈する病態(1家系において,6p21-p23に連鎖)[Melberg et al 1999]
- 運動失調,小脳萎縮,知的障害,および注意欠陥多動性障害(ADHD)の疑いを呈する病態(ナトリウムチャネル遺伝子であるSCN8Aのヘテロ接合性変異に関連)[Trudeau et al 2006]
- 遅発性(40~60歳代)の小脳性運動失調に先立って,何年も前から痙攣性咳嗽がみられた症例.1人の患者のMRIに,小脳歯状核の石灰化を認めた[Coutinho et al 2006].
分子遺伝学的検査
CAGトリプレット・リピート病. SCA1,SCA2,SCA3,SCA6,SCA7,SCA12,SCA17,およびDRPLAは当該遺伝子のコード配列内のCAG3塩基配列の過剰伸長に起因する.(CAG配列はグルタミンをコードするため,このような疾患群はポリグルタミン病とも称される)- CAG反復数に対する分子遺伝学的検査は,特異性,感受性とも高い診断法である.正常なCAG反復をもつアレルの大きさと,病原性(完全浸透)のCAG反復配列の伸長数は,疾患によって非常にばらつきがある(各疾患については,各GeneReviewを参照のこと).
CAG反復数の解釈で注意すべき2つの点: - 幾つかの疾患では,CAG反復数の正常域の上限と異常域の下限が重複しているアレルがみられる.通常,このようなアレルは易変性の正常アレル,もしくは低浸透率アレルと分類される.
易変性の正常アレル (これまで中間型アレルと呼ばれていた)易変性の正常アレルにより発症することはないが,世代ごとに伝播して低浸透率アレルとなったり,完全浸透アレルとなったりする可能性がある.このため,易変性の正常アレルをもつ者の子では,病原性アレルを受け継ぐリスクが高くなっている.
低浸透率アレル 浸透率が低いアレルにより発症する場合もあれば発症しない場合もある.このようなアレルを有する場合の確率はほとんどが不明である.
CAG反復数が易変性の正常アレルと浸透率の低下したアレルとの境界にある場合や,浸透率の低下したアレルと病原性アレルとの境界にある場合,検査結果の解釈は困難になりやすい.このような場合,臨床検査を行いつつ診察を行うと,CAG反復数の測定値の精度を判断する際に有益であると考えられる. - サザンブロット解析だけでは検出不能となることが考えられる極端に大きなCAG反復から,SCA2,SCA7,SCA8,SCA10が生じている場合がある.このような場合,明らかにホモ接合体であることを示す検査結果(PCR検査による単一アレル長のみが検出)が生じた際には,サザンブロット解析で大きなCAG伸長変異の有無を調べることが妥当であるかを判断するために,臨床所見,家族歴,発症年齢など多数の要素を考慮しつつ,検査結果を解釈しなければならない.
- その他. SCA8ではATXN8OS遺伝子にCTGの3塩基配列の反復数の伸長がみられる[Koob et al 1999].ATXN8OS遺伝子内の極度に伸長した大きな反復配列(~800)では,臨床症状が生じないことがある[Ranum et al 1999].SCA8の発症機序は複雑であり,もう1つの重複遺伝子であるATXN8遺伝子内のCAG反復も含んでいる.
SCA10ではATXN10遺伝子内にATTCTの5塩基反復の過剰伸長を認め,反復数の異常伸長の程度はCAGの反復数の異常を有する疾患でみられる場合よりもずっと大きい[Matsuura et al 2000].
表現促進現象は,症状は生じていないがリスクのある血縁者の遺伝カウンセリングや出生前診断での重要な問題である.CAG反復数の伸長と発症年齢の若年化や重症度の悪化は,一般に相関しているが,発症年齢,重症度,個別の症状,疾患の進行速度はさまざまであり,家族歴や分子遺伝学的検査では正確に予測できない.表現促進現象や3塩基配列伸長の現れに注意を払う一方で,次世代への伝播で3塩基配列の反復数が変化しなかったり,むしろ縮小したりする場合もあることに留意することが大切である.
CAG反復疾患では,伸長アレルを母親から受け継いだ場合よりも父親から受け継いだ場合に,反復数の伸長が起こりやすい.これに対してSCA8では,CTG反復数の伸長はほとんどが母親から受け継いだ場合に起こる[Koob et al 1999]
常染色体優性の小脳性運動失調症(ADCA)の臨床的特徴
常染色体優性の運動失調症の発症年齢は重複している.表2には,各病型に多少なりとも鑑別可能な臨床的特徴を記載した[Hammans 1996, Nance 1997, Scho¨ls et al 1997, Klockgether et al 1998, Kerber et al 2005, Kraft et al 2005, Maschke et al 2005].しばしば常染色体優性の失調症は臨床所見や神経画像所見から鑑別できない.こうした疾患は通常,緩徐進行性であり,また脳画像でも示されるように,しばしば小脳萎縮を伴う.表2には常染色体優性の小脳性運動失調(ADCA)における各病型の発症頻度を記した.各国から報告された常染色体優性の小脳性運動失調(ADCA)のサブタイプの頻度については,図1を参照のこと.
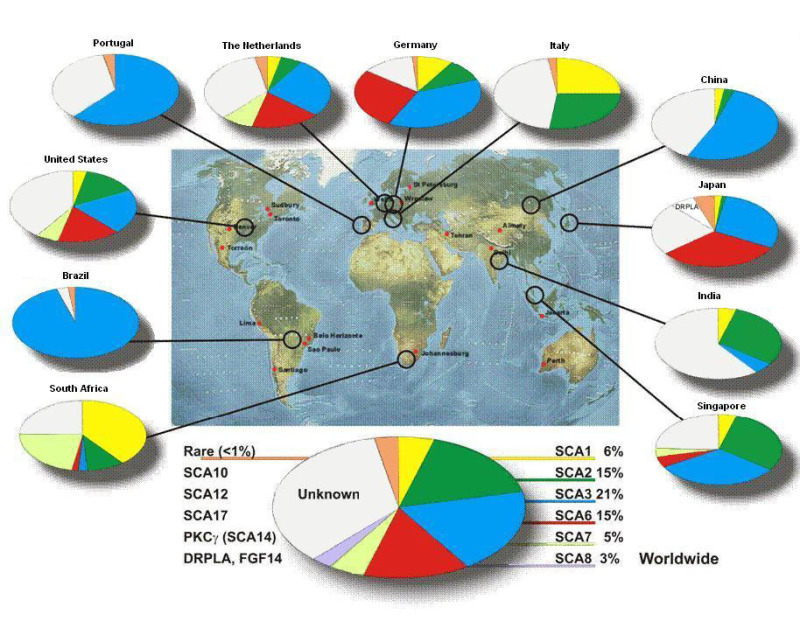
図1:SCAサブタイプの国別頻度 [Scho¨ls et al 1997, Moseley et al 1998, Saleem et al 2000, Storey et al 2000, Tang et al 2000, Maruyama et al 2002, Silveira et al 2002, van de Warrenburg et al 2002, Dryer et al 2003, Brusco et(more...)
データはMoseley et al [1998]によって行われた米国での包括的研究に基づく.遺伝性小脳性運動失調(ADCA)の各サブタイプの頻度は,創始者効果による場合が多いが,地域ごとにばらつきがあることがある.例えば,歯状核赤核淡蒼球ルイ体萎縮症 (DRPLA)は日本に多く,SCA3はポルトガルで多い.SCA2は韓国で多く,SCA3は英国よりも日本とドイツに多い[Leggo et al 1997,Scho¨ls et al 1997, Watanabe et al 1998, Kim et al 2001, Silveira et al 2002].SCA3が初めて報告されたのはアゾレス諸島出身のポルトガルの家系からであり,Machado-Joseph病(MJD)と呼ばれた.DRPLAは北米では稀であり,日本に多い.最近の研究では,日本国内でも各地で発症率にばらつきがあることがわかった[Matsumura et al 2003].表2:常染色体優性小脳失調:臨床的特徴
| 疾患名1 | 平均発症年齢(年) | 平均罹病期間(年) | 特徴的な症状 2 | その他 | 参考文献 |
|---|---|---|---|---|---|
| SCA1 | 20~30歳代 (10歳未満~60歳超) |
15年(10~28年) | 錐体路徴候,末梢ニューロパー | Subramony & Ashizawa [2011] | |
| SCA2 | 20~30歳代(10歳未満~60歳超) | 10年 (1~30年) |
衝動性眼球運動の遅延化,末梢性ニューロパチー,深部腱反射の減弱,認知症 | Pulst [2010] | |
| SCA3 | 30歳代 (10~70歳) |
10年 (1~20) |
錐体路徴候や錐体外路徴候,眼瞼後退,眼振,衝動性眼球運動の遅延化,筋萎縮による線維束性収縮,感覚消失 | Paulson [2011] | |
| SCA4 | 30~60歳代(19~72歳) | 数10年 | 感覚性軸索型ニューロパチー,難聴 | おそらく16q22に連鎖 | Flanigan et al [1996] |
| SCA5 | 20~30歳代(10~68歳) | 25年超 | 早期発症,緩徐進行性 | 初めて報告されたのはエイブラハム・リンカーンの子孫 | Ranum et al [1994], Stevanin et al [1999],Burk et al [2004], Ikeda et al [2006] |
| SCA6 | 40~50歳代(19~71歳) | 25年超 | 発作性運動失調症の場合もある.進行は非常に緩徐. | Gomez [2008] | |
| SCA7 | 20~30歳代(0.5~60歳) | 20年 (1~45年,発症年齢の低さは罹病期間の短さに相関) |
網膜症を伴う失明 | Bird et al [2007] | |
| SCA8 | 30歳代 (1~65歳) |
寿命は正常 | 緩徐進行性,深部腱反射の亢進を呈する場合もある,振動覚減弱,まれに認知障害 | Ikeda et al [2007] | |
| SCA10 | 30歳代 (12~48歳) |
9年 | 時折痙攣発作を生じる | ほとんどの家系がメキシコ系 | Matsuura & Ashizawa [2012] |
| SCA11 | 30歳 (15~70歳) |
寿命は正常 | 軽度,歩行機能は維持 | Houlden [2008] | |
| SCA12 | 30歳代 (8~62歳) |
緩徐進行性の運動失調,20歳代の運動時振戦,反射亢進,軽微なパーキンソニズムも生じうる,認知/精神障害(認知症を含む) | Margolis et al [2011] | ||
| SCA13 | 小児期,若しくは成人期 | 不明 | 軽度知的障害,低身長 | Pulst [2012] | |
| SCA14 | 20~30歳代(3~70歳) | 数10年 (1~30) |
早期発症の体幹性ミオクローヌス | Chen et al [2010] | |
| SCA15 | 30歳代 (7~66) |
数10年 | 純粋型の運動失調,進行は非常に緩徐 | Storey [2011] | |
| SCA16 | 39歳 (20~66歳) |
1~40年 | 頭部振戦 | 日本人1家系 | Miyoshi et al [2001], Miura et al [2006] |
| SCA17 | 30歳代 (3~55歳) |
8年超 | 精神機能低下.舞踏病,ジストニー,ミオクローヌス,てんかんが生じることもある. | プルキンエ細胞消失,伸長したポリグルタミンの核内封入体 | Toyoshima et al [2012] |
| SCA18 | 青年期 (12~25歳) |
数10年間 | 初期の感覚/運動ニューロパチー,眼振,構語障害,腱反射減弱 | 筋脱力,萎縮,筋束性収縮,バビンスキー反射 | Brkanac et al [2002], Brkanac et al [2009] |
| SCA19/22 | 30歳代 (10~51歳) |
数10年間 | 緩徐進行性,認知障害はまれ,ミオクローヌス,反射亢進 | 9家系 | Schelhaas et al [2001], Verbeek et al [2002],Chung et al [2003], Lee et al [2012] |
| SCA20 | 40歳代 (19~64歳) |
数10年間 | 初期の構語障害,痙攣性発声障害,反射亢進,運動緩慢 | 歯状核の石灰化 | Storey [2012] |
| SCA21 | (6~30歳) | 数10年間 | 軽度認知障害 | Devos et al [2001] | |
| SCA23 | 40~50歳代 | 10年超 | 構語障害,眼球運動異常,振動覚と位置覚の減弱 | オランダ人1家系.神経病理学的所見3 | Verbeek et al [2004] |
| SCA25 | (1.5~39歳) | 不明 | 感覚ニューロパチー | フランス人1家系 | Stevanin et al [2003] |
| SCA26 | (26~60歳) | 不明 | 構語障害,追跡眼球運動異常 | ノルウェー系アメリカ人1家系.MRI:小脳萎縮 | Yu et al [2005], Hekman et al [2012] |
| SCA27 | 11歳 (7~20歳) |
数10年 | 若年発症の振戦.ジスキネジー,認知障害 | オランダ人1家系 | van Swieten et al [2003], Brusse et al [2006] |
| SCA28 | 19.5歳 (12~36歳) |
数10年 | 眼振,眼筋麻痺,眼瞼下垂,腱反射亢進 | イタリア人2家系 | Cagnoli et al [2006], Mariotti et al [2008],Edener et al [2010] |
| SCA29 | 小児初期 | 生涯を通じて | 学習障害 | Dudding et al [2004] | |
| SCA30 | (45~76歳) | 生涯を通じて | 反射亢進 | Storey et al [2009] | |
| SCA31 | 40~50歳代 | 生涯を通じて | 感覚は正常 | Nagaoka et al [2000] | |
| SCA35 | 43.7+/-2.9(40~48)歳 | 15.9+/-8.8 (5~31)歳 |
反射亢進, バビンスキー反射 |
痙性斜頚 | Wang et al [2010] |
| SCA36 | 52.8+/- 4.3歳 | 数10年 | 筋束性収縮,舌萎縮,反射亢進 | Kobayashi et al [2011] | |
| DRPLA | 20~30歳代 (8~20歳,もしくは40~60歳代) |
発症年齢の低さは罹病期間の短さに相関 | 舞踏病,痙攣発作,認知症,ミオクローヌス | ハンチントン病と混同される場合が多い. | Tsuji [2010] |
| EA1 | 生後から10歳代(2~15歳) | 20歳以降は減弱 | ミオキミア.持続時間は数秒から数分.驚愕もしくは運動により生じる.回転性めまいは生じない. | D'Adamo et al [2012] | |
| EA2 | (2~32歳) | 生涯を通じて | 眼振.持続時間は数分から数時間.姿勢の変化により生じる.その後,永続的な運動失調となる. | Spacey [2011] | |
| SPAX1 | (10~20歳) | 寿命は正常 | はじめは進行性の下肢痙直 | ARSACS(常染色体劣性のシャルルボア‐サギュネー[Charlevoix-Saguenay]型痙性失調症)に類似 |
ADCA =遺伝性小脳性運動失調
SCA =脊髄小脳失調症
DRPLA =歯状核赤核淡蒼球ルイ体萎縮症
SAX =痙性運動失調
EA =発作性運動失調症
DTRs =深部腱反射
SPAX1 =常染色体優性痙性運動失調1型
- SCA9は割り当てられていない.
- すべてに失調性歩行がみられる.
- プルキンエ細胞の喪失,脊髄後柱と側柱における脱髄化,黒質における神経細胞核内封入体
常染色体劣性の遺伝性運動失調症
運動失調を呈する常染色体劣性疾患をまとめた(Embirucu et al [2009]のレビューを参照).
表3と表4には,運動失調を主症状とする11の常染色体劣性疾患に関する情報をまとめた.これらの疾患は現時点で劣性遺伝性の運動失調症に関する分子遺伝学的な知見がどの程度まで集積されているかを示すために選んだものである.その他の稀な常染色体劣性遺伝性運動失調症については簡潔に記載する.
常染色体劣性遺伝性運動失調症の分子遺伝学
表3:さまざまな常染色体劣性の遺伝性運動失調症:分子遺伝学
| 疾患名 | 遺伝子記号/ 蛋白名 |
参考文献 | 検査の 実施状況 |
|---|---|---|---|
| Friedreich失調症 (FRDA) | FXN / frataxin | Bidichandani & Delatycki [2012] | 臨床 |
| 毛細血管拡張性運動失調症(ataxia-telangiectasia) (A-T) | ATM | Gatti [2010] | 臨床 |
| ビタミンE単独欠乏失調症(ataxia with vitamin E deficiency) (AVED) | TTPA | Schuelke [2010] | 臨床 |
| 眼球運動失行を伴う失調症(ataxia with oculomotor apraxia type 1) (AOA1) | APTX / aprataxin | Coutinho & Barbot [2010] | 臨床 |
| 眼球運動失行を伴う失調症(ataxia with oculomotor apraxia type 2) (AOA2) | SETX | Moreira & Koenig [2011] | 臨床 |
| IOSCA 1) | C10orf2 / twinkle | Nikali & Lo¨nnqvist [2010] | 臨床 |
| Marinesco-Sjo¨gren症候群 | SIL1 | Anttonen & Lehesjoki [2010] | 臨床 |
| 常染色体劣性Charlevoix-Saguenay型痙性失調症 (ARSACS) | SACS / sacsin | Vermeer et al [2012] | 臨床 |
| Refsum病 | PHYH PEX7 |
Wanders et al [2010] | 臨床 |
| CoQ10 欠乏症 | CABC1 COQ2 COQ9 PDSS1 PDSS2 |
Montero et al [2007] | 臨床 |
| 脳腱黄色腫症(CTX) | CYP27A1 | Federico et al [2011] | 臨床 |
- IOSCA =乳児期発症脊髄小脳失調症
常染色体劣性遺伝性運動失調症の臨床的特徴
表4. 常染色体優性小脳失調:臨床的特徴
| 疾患名 | 発症頻度 | 発症年齢
|
罹病期間(年) | 特徴的な症状 |
|---|---|---|---|---|
| Friedreich失調症 (FRDA) | 1~2:50,000 | 生後~10歳代(4~40歳) | 10~30年間 | 反射減弱,バビンスキー反射,感覚消失,心筋症 |
| 毛細血管拡張性運動失調症(ataxia-telangiectasia) (A-T) | 1:40,000~100,000 | 生後~10歳まで | 10~20年間 | 毛細血管拡張症,免疫不全,癌,染色体不安定性,αフェトプロテイン増加 |
| ビタミンE単独欠乏失調症(ataxia with vitamin E deficiency) (AVED) | まれ | 2~52歳(通常20歳未満) | 数10年間 | フリードライヒ運動失調症と類似,頭部動揺(28%) |
| 眼球運動失行を伴う失調症(ataxia with oculomotor apraxia type 1) (AOA1) | 不明 | 小児期 | 数10年間 | 眼球運動失行,舞踏病アテトーゼ,軽度知的障害,低アルブミン血症 |
| 眼球運動失行を伴う失調症(ataxia with oculomotor apraxia type 2) (AOA2) | 不明 | 10~22歳 | 数10年間 | 小脳萎縮,感覚運動ニューロパチー,眼球運動失行 |
| IOSCA 1) | まれ(フィンランド) | 幼児期 | 数10年間 | 末梢性ニューロパチー,アテトーゼ,視神経萎縮,難聴,眼筋麻痺 |
| Marinesco-Sjo¨gren症候群 | まれ | 幼児期 | 数10年間 | 知的障害,白内障,筋緊張低下,筋障害 |
| 常染色体劣性Charlevoix-Saguenay型痙性失調症 (ARSACS) | まれ | 小児期 | 数10年間 | 痙直,末梢性ニューロパチー,線状網膜(retinal striation) |
| Refsum病 | まれ | 生後~50歳代 | 数10年間 | ニューロパチー,難聴,魚鱗癬,網膜症 |
| CoQ10 欠乏症 | まれ | 小児期 | 数10年間 | 発作,認知機能低下,錐体路徴候,筋障害 |
| 脳腱黄色腫症(CTX) | 1:50,000 | 小児期から成人初期 | 数10年間 | 肥厚した腱(thick tendons),認知機能低下,ジストニー,白質病変,白内障 |
フリードライヒ運動失調症(FRDA)の特徴は緩徐進行性の運動失調であり,通常25歳までに発症する.通常,腱反射の減弱,構語障害,バビンスキー反射,位置感と振動感の喪失を呈する[Lynch et al 2006].患者の約25%では発症年齢が遅くなったり(25歳超),腱反射が維持されたり,進行が例外的に緩徐であるといった「非定型」の特徴がみられることがある.たいていの患者はFXN遺伝子のGAA3塩基反復の過剰伸長が見られる.CAGの3塩基反復によって生じる常染色体優性小脳性運動失調と異なり,フリードライヒ運動失調症で表現促進現象は生じない[Durr et al 1996].
毛細血管拡張性小脳失調症(A-T)の特徴は,1~4歳に発症する進行性小脳性運動失調,眼球運動失行,易感染性,舞踏病アテトーゼ,結膜の毛細血管拡張症,免疫不全症,悪性腫瘍リスクの増加(特に白血病とリンパ腫)である.毛細血管拡張性小脳失調症(A-T)の診断を支持する検査所見は,末梢血の核型分析で7;14染色体の転座が同定されること,免疫不全症がみられること,in vitroでの放射線感受性試験などがある.ATM遺伝子の分子遺伝学的検査も行われている.
ビタミンE単独欠乏失調症(AVED)は通常,学童期から10歳代に構語障害や,(特に暗所での)歩行時の平衡障害を伴って発症する.早期からの固有感覚の欠如により不器用が進行する.ジストニア,精神病エピソード(妄想症),色素性網膜症,知能低下が見られることもある.大半の患者は運動失調と下肢の筋力低下のために11歳~50歳の間に車椅子生活となる.臨床的にフリードライヒ運動失調症と似ているが,ビタミンE欠乏を伴う運動失調(AVED)ではフリードライヒ運動失調症と比べて頭部動揺やジストニーを伴うことが多く,心筋症を呈することは少ない.ビタミンE欠乏を伴う運動失調(AVED)はビタミンEの補給により治療できるので,(血清ビタミンEの濃度を調べて)AVEDかどうかを見極めることが重要である[Yokota et al 1997,Cavalier et al 1998].
ビタミンE欠乏を伴う常染色体劣性の運動失調(AVED)だけの患者で期待されるのとは異なり,SCA8と合併している患者では,ビタミンEの補充は奏効しなかった[Cellini et al 2002].
グランドケイマン島でみられる別の常染色体劣性運動失調は,ビタミンEの代謝にも関与するCRAL-TRIO蛋白をコードする遺伝子であるATCAY遺伝子変異が原因である[Bomar et al 2003].
眼球運動失行を伴う失調症1型(AOA1)の特徴は,小児発症(平均発症年齢~7歳)の緩徐進行性の小脳性運動失調であり,数年後に眼球運動失行が生じ外眼筋麻痺へと進行する.すべての患者には重度の原発性末梢性運動ニューロパチーが生じ,発症から約7~10年後に四肢麻痺に至り,歩行機能を喪失する.ポルトガルに祖先を持つ患者では知能は正常のままであるが,日本人患者では精神機能の悪化が生じる.眼球運動失行を伴う運動失調1型(AOA1)の診断は臨床所見に基づいて行われ,分子遺伝学的検査で確定される[Barbot et al 2001, Date et al 2001, Moreira et al 2001, Le Ber et al 2003, Onodera 2006].
眼球運動失行を伴う失調症2型(AOA2)は,発症年齢が10~22歳であり,小脳萎縮,軸索型感覚運動ニューロパチー,眼球運動失行,αフェトプロテインの血清濃度の上昇を特徴とする[Moreira et al 2003, Asaka et al 2006].眼球運動失行を伴う運動失調2型(AOA2)は臨床検査と生化学検査の結果や家族歴に基づき,毛細血管拡張性小脳失調症と眼球運動失行を伴う運動失調1型(AOA1)との鑑別が必要であり,分子遺伝学的検査に基づいて確定される.
乳児期発症脊髄小脳失調症(IOSCA)はフィンランドから報告された稀な疾患であり,小脳,脊髄,および脳幹の変性と,軸索型の感覚ニューロパチーを伴う[Nikali et al 2005].
マリネスコ-シェーグレン(Marinesco-Sjo¨gren)症候群 は,運動失調に知的障害,白内障,低身長,筋緊張低下が併発する稀な病型である[Zimmer et al 1992, Anttonen et al 2005, Senderek et al 2005].
常染色体劣性シャルルボア‐サギュネー(Charlevoix-Saguenay)型痙性運動失調症(ARSACS)は,若年発症(12~18ヶ月)の歩行困難と不安定歩行を特徴とする.主な神経徴候は運動失調,構語障害,痙直,伸展性足底反射,遠位筋の消耗,下肢優位の感覚運動ニューロパチー,水平方向の注視眼振であり,ほぼすべてが進行性である.ケベック州のシャルルボア‐サギュネー(Charlevoix-Saguenay)型痙性失調症(ARSACS)家系の患者の網膜では,過剰にミエリンが形成された線維が,眼底の辺縁部から放射線状に伸びている[Bouchard et al 1998].こうした網膜の変化は,フランス系,チュニジア系,トルコ系のARSACS患者では稀である[Mrissa et al 2000, Pulst & Filla 2000].ARSACS患者が車椅子生活となるのは平均して41歳である.認知機能は長期間,維持され,患者は成人後期になるまで日常生活動作を遂行できる.死亡年齢は通常,50歳代である.
レフサム病の発症は一般に小児期から成人初期であり,運動失調に加えて末梢性ニューロパチー,難聴,魚鱗癬,もしくは色素性網膜炎が生じる[Wanders et al 2010].
PHARC(多発ニューロパチー,難聴,運動失調,色素性網膜炎,白内障)は,ABHD12遺伝子の変異から発症する[Fiskerstrand 2010].
補酵素Q10欠乏が発作,認知機能の低下,錐体路徴候,ミオパチーを伴うことは多いが,顕著な小脳性運動失調を併発することもある[Musumeci et al 2001, Lamperti et al 2003, Montero et al 2007].症状は補酵素Q治療が奏効する場合がある.
脳腱黄色腫症(CTX)では特徴的な腱の肥厚が見られる.認知機能の低下,ジストニー,白内障,脳MRI上の白質病変を伴う場合が多い.
遺伝子変異は同定されているが(表3・表4に含まれていない)このほかの常染色体劣性の小脳性運動失調を以下に掲げる.
- 小脳萎縮,運動失調,軸索型感覚運動ニューロパチーの見られるサウジアラビア人の1家系(14番染色体q31-q32に連鎖,トポイソメラーゼ1依存性DNA損傷修復酵素をコードするTDP1遺伝子変異に連鎖,SCAN1)[Takashima et al 2002, Walton et al 2010](「軸索型ニューロパチーを伴う常染色体劣性の脊髄小脳失調症」を参照).
- てんかんや認知障害を伴うこともある小児期発症の運動失調を呈する血族婚のサウジアラビアの1大家系では,(rundataxinをコードする)K1AA0226遺伝子のフレームシフト変異が見られる[Assoum et al 2010].
- 小脳下部の低形成と脳回の軽度の単純化がみられる非進行性の小脳性運動失調と知的障害がみられるフッター派家系では,(超低比重リポ蛋白受容体をコードする)VLDLR遺伝子の変異が見られる[Boycott et al 2005](「VLDLR関連小脳低形成」を参照).
- 遅発性の小脳性運動失調の見られるケベックのボース地区の幾つかのフランス-カナダ系家系では,SYNE1遺伝子変異に関連する[Gros-Louis et al 2007, Dupre et al 2007](「SYNE1関連疾患」を参照).
- 程度は様々な先天性小脳無形成/低形成を伴う疾患
- MRIで橋部に臼歯状の徴候が示されるジュベール症候群[Dixon-Salazar et al 2004, Ferland et al 2004]
- 新生児糖尿病を伴う小脳無形成[Sellick et al 2004]
- 超低比重リポ蛋白受容体(VLDLR)関連小脳低形成
- 先天性グリコシル化障害[Grunewald et al 2002]
- 橋小脳低形成[Renbaum et al 2009, Cassandrini et al 2010]
- 血族婚の日本人同胞では,小児期発症の精神運動遅滞が生じた後,成人発症の失調性歩行が生じたが,MRIには小脳虫部萎縮を認め,(シナプトタグミン -14をコードする)SYT14遺伝子のミスセンス変異をホモ接合で認めた[Doi et al 2011].
- 常染色体劣性脊髄小脳失調症9型(SCAR9)Lagier-Tourenne et al [2008]は血族婚のアルジェリア系家系について報告を行い,数人の小児期発症の進行性小脳性運動失調患者に,軽度精神運動遅滞を伴う場合があったことを報告した.補酵素Q10の生合成に関与するADCK3遺伝子(CABC1)に,ホモ接合性のスプライス部位変異が見つかった.
- Vermeer et al [2010]は,失調性歩行,下向眼振,構語障害,腱反射亢進,及びMRIでの小脳萎縮所見を呈する青年期から成人初期に発症する常染色体劣性の運動失調を報告した.筋電図で下位運動ニューロン疾患の筋束性収縮を認める場合もある.アノクタミン-10をコードするANO10遺伝子にホモ接合やヘテロ接合の変異が検出された.
- Emre Onat et al [2012]は,ATP8A2遺伝子のミスセンス変異(p.Ile376Met)に伴う運動失調,知的障害,四足歩行のみられる血族婚のトルコ系家系を報告した.
- Guergueltcheva et al [2012]は,代謝調節型グルタミン酸受容体1をコードするGRM1遺伝子変異から生じるローマ人における常染色体劣性の先天性小脳性運動失調を報告した.患者には運動失調に加えて発達遅滞や知的障害が生じたり,器質的に脳が小さくなったりすることがある.
このほか,まだ遺伝子変異が同定されていない,(表3や表4に含まれない)常染色体劣性の小脳性運動失調を以下に掲げる.
- 日本で報告された知的障害,末梢性ニューロパチー,及び顕著な小脳萎縮を伴う運動失調[Tachi et al 2000].
- 脊髄後柱の変性と色素性網膜炎を伴う運動失調[Higgins et al 1997]
- 低性腺刺激ホルモン性性腺機能低下症を伴う運動失調 類似の同胞群では,コエンザイムQ10欠乏を認めた[Gironi et al 2003].
- 血族婚のレバノン系家系での知的障害,視神経萎縮,皮膚病変を伴う運動失調(15q24-q26に連鎖)[Megarbane et al 2001, Delague et al 2002]
- 難聴と視神経萎縮を伴う運動失調(6p21-p23に連鎖)[Bomont et al 2000]
- 14人の同胞中5人が運動失調,衝動性眼球運動の混入,感覚ニューロパチー,ミオクローヌスを呈する単独のスロヴェニア家系[Swartz et al 2002]
- 血族婚のノルウェーの大家系における幼児期発症の非進行性運動失調(20q11-q13に連鎖)[Tranebjaerg et al 2003]
- ビオチニダーゼ欠乏症の年長児に,しばしば運動失調と発達遅滞が見られる.
- MRと小脳萎縮を呈するパレスチナ家系(22q11に連鎖)[Baris et al 2005]
X連鎖性遺伝性運動失調症
X連鎖性鉄芽球性貧血を伴う運動失調(XLSA/A)の特徴は,若年発症の運動失調,測定障害,及び反復拮抗運動不全である.運動失調は非進行性である場合もあれば緩徐進行性である場合もある.上位運動ニューロン徴候(深部腱反射の亢進,非持続性の足クローヌス,伸展性足底反応は不定もしくは陽性)を呈する男性もいる.軽度の学習障害が生じる.貧血は軽度であり,症状はない.保因者女性の神経学的検査は正常である.病原性変異はミトコンドリアの鉄輸送に関与する蛋白をコードするABC7遺伝子にあり,フリードライヒ運動失調症と共通した発症機序が想定される[Allikmets et al 1999, Bekri et al 2000, Maguire et al 2001].
とりわけ男性では,成人発症の運動失調は,脆弱X随伴振戦/失調症候群(FXTAS)の一部である可能性がある[Berry-Kravis et al 2007, Leehey 2009](「FMR1関連疾患」を参照).
ミトコンドリア病を伴う運動失調
進行性の運動失調に,MERRF(赤色ぼろ線維・ミオクローヌスてんかん症候群),NARP症候群(ニューロパチー・運動失調症・色素性網膜炎)[Finsterer 2009b],カーンズ・セイヤー(Kerns-Sayre)症候群などのミトコンドリア病が併発することがある.ミトコンドリア病では発作,難聴,糖尿病,心筋症,網膜症,低身長など他の臨床徴候を伴うことが多い[Da Pozzo et al 2009].
Pfeffer et al [2012]は,MTATP6遺伝子のミスセンス変異が眼球運動異常,構語障害,脱力,軸索ニューロパチー,反射亢進を伴うこともある小児期発症/成人発症の小脳性運動失調の原因となる可能性を報告した.評価手順
ある個人に遺伝性運動失調症が考えられる場合,予後や遺伝カウンセリングの話し合いで一助となる特異的原因を確定する際には,以下の手順を用いることができる.ある個人での遺伝性運動失調症の特異的原因を確定する際には,通常,病歴聴取,身体診察,神経学的検査,神経画像診断,詳細な家族歴の聴取,分子遺伝学的検査が行われる.
臨床所見.
遺伝性運動失調ではすべてのタイプの臨床所見が広範に重複しているため,運動失調を呈するある個人に常染色体優性の遺伝形式に一致する家族歴があっても,分子遺伝学的検査を行うことなく診断を確立するのは困難である.臨床所見は常染色体劣性の運動失調の幾つかを鑑別する際に役立つことがある.
家族歴.
神経学的徴候や症状がみられない血縁者に注意を払って,3世代の家族歴を聴取すること.血縁者の関連所見の記録は,該当者に直接検査を行って入手することもできるが,分子遺伝学的検査,神経画像診断,剖検記録など,該当者の医療記録を読み返すことからも入手可能である.
検査.
DNAを用いない臨床検査を行うことができるのは,常染色体劣性の遺伝性運動失調症である毛細血管拡張性小脳失調症(A-T)とビタミンE欠乏を伴う運動失調(AVED)の2疾患である.
分子遺伝学的検査.
Gasser et al [2010]は,DNA解析を用いた臨床診断の検査手順について論じた.
家族歴が常染色体優性遺伝を示唆している場合の検査手順
- 常染色体優性の遺伝性運動失調症の推定50~60%(表1参照)は,SCA1,SCA2,SCA3,SCA6,SCA7,SCA8,SCA10,SCA12,SCA17,DRPLAに対して精度と特異性の高い分子遺伝学的検査を行うことから確定できる.すべて,該当する遺伝子において,3塩基配列の反復配列の伸長を認める.
- 臨床所見が広範に重複していることから,遺伝性運動失調症の検査を行う検査機関のほとんどでは,SCA1,SCA2,SCA3,SCA6,SCA7,SCA10,SCA12,SCA14,SCA17への検査を含む一連の検査を行っている.疾患頻度に基づいて2段階の検査方式を行っている検査機関が多く,まず,発症頻度の高い運動失調であるSCA1,SCA2,SCA3,SCA6,SCA7が検査される.
- 反復配列の伸長の生じない幾つかの常染色体優性のSCA(SCA5,SCA13,SCA14,SCA27,16q22連鎖SCA)に対しても,検査が可能である.
- 疾患頻度の低い遺伝性失調症については,個別に検査を行うべきである.民族的背景(例外[Fujigasaki et al 2002, Matsuura et al 2002]もあるが,メキシコ系に対してはSCA10の検査)や,発作がみられる場合にはSCA10を,振戦がみられる場合にはSCA12を,認知障害や舞踏病がみられる場合にはSCA17を,経過の長い純粋小脳型ではSCA6,SCA8,SCA14を,といったように,特定の因子に基づいて検査を行ってもよい.
- 検査から特定の疾患が強く示唆される場合,(たとえば網膜症の存在からSCA7が疑われる場合),あるいは特定の疾患の家族歴が見られる場合,1つの疾患に対してのみ検査を行えばよい.
- 注意しなければならない点は,(1)これらの疾患の多くで,CAG反復数を異常とする範囲が正確に確立されている訳ではないこと,また(2)SCA8についてはごく少数の家系しか報告されていないことから,浸透率や性差による影響が完全に解明されていないため,検査結果の解釈が複雑になることがある[Gupta & Jankovic 2009].このため,このような検査を受ける患者への診断や遺伝カウンセリングでは,経験のある臨床検査技師,遺伝専門医,遺伝カウンセラーによる支援が必要である.
- 運動失調に対する一連の検査にかかる費用は,MRI検査と同程度になることが多いことにも注意を要する.分子遺伝学的検査での陽性結果は,遺伝性運動失調症のMRI所見よりも特異度が高い.運動失調症の家族歴がない患者では,分子遺伝学的検査の結果が陽性となる確率は低いが,通常こうした検査の実施は,医学的評価や遺伝カウンセリングに際して特異的診断を確立する目的で妥当とみなされる.
家族歴が常染色体劣性遺伝を示唆している場合の検査手順
(罹患者が同胞のみである場合や,両親が近親婚である場合など)
.家族歴で,罹患者が同胞のみである場合や,両親が近親婚である場合には,常染色体劣性の遺伝形式が考えられる.発症頻度や治療の可能性を考えると,フリードライヒ運動失調症,毛細血管拡張性小脳失調症,ビタミンE単独欠乏失調症,レフサム病や慢性あるいは成人発症のヘキソサミニダーゼA欠損症(GM2ガングリオシドーシス)のような代謝異常や脂質蓄積症を考慮すべきである.
患者が孤発例となっている場合の検査手順
(すなわち,時に誤って「散発」症例とされる家系内で唯一の発症である場合).
運動失調に明らかな後天的原因が見出せない場合,患者がSCA1,SCA2,SCA3,SCA6,SCA8,SCA17,フリードライヒ運動失調症のいずれかである可能性は約13%である[Abele et al 2002].このほかの可能性として考慮すべき点は,別の常染色体優性の運動失調に生じた新生突然変異,浸透率の低下,父親が異なる場合,もしくは常染色体劣性やX連鎖性の家系での唯一の発症例である場合である.
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
遺伝性運動失調症の遺伝形式は,常染色体優性,常染色体劣性,X連鎖劣性である.発端者に運動失調に関連する特異的症状がみられる場合(たとえば,ミトコンドリア病や脆弱X随伴振戦/失調症候群[FXTAS]で運動失調を認める場合など),当該疾患に対するカウンセリングの適応となる.
血縁者のリスク-常染色体優性の遺伝性運動失調症
発端者の両親
- 家族歴が陰性の場合も時折みられるが,ほとんどの場合,常染色体優性の運動失調と診断された患者の両親のどちらかは罹患者である.
- 親が早く死亡した場合,家系内の常染色体優性の運動失調を認識していない場合,親の発症が遅い場合,浸透率が低下したため異常アレルを有する親が無症状のままでいる場合,もしくは新生突然変異である場合,家族歴が陰性にみえることがある.
発端者の同胞
- 同胞のリスクは発端者の両親の遺伝学的状態によって異なる.
- 発端者の両親の1人が変異アレルを有する場合,同胞が変異アレルを受け継ぐリスクは50%である.
発端者の子
常染色体優性の運動失調の患者が子に変異アレルを伝える確率は50%である.
血縁者のリスク-常染色体劣性の遺伝性運動失調症
発端者の両親
- 両親は絶対的ヘテロ接合体であるため,病原性遺伝子変異を1つ有する.
- ヘテロ接合体に症状は生じない.
発端者の同胞
- 受精時点で,発端者の子が罹患する確率は25%,無症状の保因者となる確率は50%,罹患者とも保因者ともならない確率は25%である.
- リスクのある同胞が罹患していないことがわかった場合,この同胞が保因者である確率は2/3となる.
発端者の子
すべての子は絶対的保因者である.
血縁者のリスク-X連鎖性遺伝性運動失調症
発端者の両親
- 男性罹患者の父親は発症することも,変異の保因者であることもない.
- 息子が罹患者であったり,血縁男性に罹患者がいる女性は,絶対的ヘテロ接合体である.
発端者の同胞
- 発端者のリスクは母親の保因者状態によって異なる.
- 発端者の母親が病原性遺伝子変異を有する場合,各妊娠で変異が受け継がれる確率は50%である.変異を受け継いだ男性同胞は発症する.変異を受け継いだ女性同胞は保因者となるが,通常,症状が現れることはない.
- 一見,発端者が孤発例(家系内で唯一の発症)であるようにみえる場合や,母親の白血球DNAに病原性遺伝子変異が検出されない場合,同胞のリスクは低いが,母系の生殖細胞系モザイクの可能性があるため,一般人口のリスクよりは高い.
発端者の子.
男性罹患者の娘は必ず保因者となるが,息子は罹患しない.
遺伝カウンセリングに関連した問題
発症リスクがあるが,まだ症状が現れていない成人血縁者への検査
発症リスクがあるが,まだ症状が現れていない常染色体優性の小脳性運動失調を有する患者の成人血縁者への検査は,分子遺伝学的検査で疾患のタイプと家系内変異が確定している場合には可能である.このような検査は,正式な遺伝カウンセリングのなかで行うべきである.この検査は,症状が現れていない患者の発症年齢や重症度,症状のタイプ,進行速度を予測するには有用でない.発症リスクがあるが症状が現れていない個人に,非特異的な症状や境界域にある症状が生じた際に行われる検査は予測的検査であり,診断検査ではない. 発症リスクのある個人に対して検査を行う場合,現在利用可能な技法で変異が同定可能であるかを確かめるため,まず家系内の罹患者の検査を行うべきである.常染色体優性の運動失調の発症リスクがあるが症状が現れていない29人に対する検査の結果が報告されている[Goizet et al 2002].
18歳未満の無症状者に対する検査
治療法がない成人発症の疾患のリスクがある18歳未満の無症状者に対して検査を実施することは不適切だとみなされている.その主たる理由は,検査による実質的な利益が期待できないにもかかわらず子どもの自律の原則を奪う結果になるからである.また,そのような情報が家族関係に悪影響を及ぼす可能性,将来的に遺伝学的な差別を受ける危険性,遺伝学的な情報を知ることにより不安が生じる可能性などの懸念も存在する.
詳しい情報については,子どもに対する遺伝学的検査に関する米国遺伝カウンセラー学会の決議文も参照のこと.米国人類遺伝学会と米国臨床遺伝学会は考慮すべき点として,小児期および青年期における遺伝学的検査の倫理的,法的,心理的影響を挙げている.
DNAバンキング は,将来の使用のために,通常は白血球から調整したDNAを貯蔵しておくことである.検査手法や,遺伝子,変異,疾患への理解は将来改善する可能性があり,患者のDNAを貯蔵しておくことは考慮されるべきである.DNAバンキングを行っている機関一覧を参照のこと.
出生前診断
遺伝性運動失調症のいくつかのタイプに対する出生前診断は,(妊娠10~12週頃に実施される絨毛生検(CVS)や妊娠15~18週頃に実施される羊水穿刺で採取された)胎児のDNAの病原性遺伝子変異に対する解析により可能である.出生前診断の実施前に,家系内患者の病原性アレルが同定されていることが条件である.
注:妊娠週数とは最終月経の第1日から換算するか,超音波検査による計測によって算出される.
(多くが)成人発症である疾患に対する出生前診断の要望は多くない.出生前診断の利用に関しては,特に出生前診断の目的が早期診断ではなく中絶が検討されて場合,医療従事者内でも家族内でもさまざまな意見があるだろう.出生前診断の決定を両親の選択として捉えている医療機関がほとんどであるが,これらの問題について慎重な議論を行うことが望ましい.
着床前診断(PGD)は病原性遺伝子変異が同定されている家系で可能である.PGDを提供している施設に関してはこちらを参照.
注:GeneTests Laboratory Directoryに掲載されている検査機関で検査が臨床的に検査が行われている場合に限り,臨床的に実施されているとするのがGeneReviewsの方針である.こうした掲載には著者,編集者,査読者の意向は必ずしも反映されていない.
臨床的マネジメント
症状の治療
運動失調への臨床的マネジメントは,通常,リハビリテーション医学と,作業療法,および理学療法で確立された方法論を基にして,協調運動障害に対する支援を行うことに向けられる.
- 杖,歩行器,車椅子は失調性歩行に有効である.
- 筆記,ボタンをかける動作,食器の使用を支援する特殊な器具が開発されている.
- 言語療法は構語障害を呈する患者に有効なことがある.重度の言語障害を呈する患者には,コンピューターを用いたコミュニケーション機器が有用である.
一次病変の予防
ビタミンE欠乏を伴う運動失調(AVED)でのビタミンE療法を除いては,遺伝性運動失調症に特異的な治療法は存在しない.
リスクの高い血縁者の検査
遺伝カウンセリング目的のリスクのある親族に対する検査に関連する問題は,「遺伝カウンセリング」を参照のこと.
研究中の治療
Underwood & Rubinsztein [2008]は,3塩基配列の反復配列の伸長を伴う運動失調の治療として有望な治療戦略に関するレビューを行った.
さまざまな疾患や病態に対する臨床試験に関する情報へアクセスしたい場合には, ClinicalTrials.govを参照のこと.注:当該疾患に対する臨床試験が行われていない場合もある.
患者登録
任意の患者登録への連絡先に関しては,GeneReviewsスタッフにお問い合わせください.
National Ataxia Registry(米国)
電話: 352-273-9194
ファックス: 352-392-8058
電子メール: nationalataxiaregistry@neurology.ufl.edu
ウェブ: www.nationalataxiaregistry.org
その他
遺伝クリニックは,患者や家族に自然経過,治療,遺伝形式,患者家族の遺伝的発症リスクに関する情報を提供とするとともに,患者の立場からの情報も提供する.Gene Test Clinic Directoryを参照のこと.
患者情報 本疾患の支援グループや複数疾患にまたがった支援グループについては「患者情報」を参照のこと.これらの機関は患者やその家族に情報,支援,他の患者との交流の場を提供するために設立された.
参考情報
患者情報 本疾患の支援グループや複数疾患にまたがった支援グループについては「患者情報」を参照のこと.これらの機関は患者やその家族に情報,支援,他の患者との交流の場を提供するために設立された.遺伝クリニックは,患者や家族に自然経過,治療,遺伝形式,患者家族の遺伝的発症リスクに関する情報を提供とするとともに,患者サイドに立った情報も提供する.
更新履歴
- Gene Review著者: Thomas D. Bird, MD
日本語訳者: 吉田邦広(信州大学医学部脳神経内科,リウマチ・膠原病内科)
Gene Review 最終更新日: 2006.4.27. 日本語訳最終更新日: 2006.8.27 - Gene Review著者: Thomas D Bird, MD
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: 2012.11.1. 日本語訳最終更新日: 2013.5.3. (in present)