先天性副腎過形成 21水酸化酵素欠損症
(21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia)
[21-OHD; CAH, 21-OHD; Virilizing Adrenal Hyperplasia]
Gene Review著者:Maria I New, MD ; Saroj Nimkarn, MD
日本語訳者: 塚本幸子,臼井健 (国立病院機構京都医療センター)
Gene Review 最終更新日: 2006.9.7. 本語訳最終更新日: 2007.9.21
原文 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia
要約
疾患の特徴
21水酸化酵素欠損症(21-OHD)は先天性副腎過形成(CAH)のなかで最も頻度の高い疾患であり,副腎皮質におけるコレステロールからコルチゾールへの合成障害を認める常染色体劣性遺伝疾患である.21-OHD CAHでは,副腎アンドロゲンの過剰な生合成によりすべての患者に男性化を認め,また一部の患者に塩類喪失を認める.重症の酵素欠損を認め出生前に発症する古典型と,軽度の酵素欠損を認め出生後に発症する非古典型とに区別される.古典型はさらに単純男性化型(〜25%)とアルドステロン産生が不十分な塩類喪失型(75%以上)に分けられる.塩類喪失型の21-OHDの新生児は生命を脅かす塩類喪失クリーゼのリスクがある.非古典型の患者は中等度の酵素欠損のみであり,出生後に高アンドロゲン血症を示す;非古典型の女性は出生時には男性化は認めない.
診断・検査
21-OHDの診断:以下の場合,本疾患を疑う.
- 出生時に男性化を認める女児,出生後に男性化を呈する女児,
- 思春期早発や副腎皮質性思春期徴候を認める女児
- 小児期に男性化を認める男児
- 性別を問わず生後4週以内に塩類喪失クリーゼを伴う新生児
治療
古典型21-OHDの治療はグルココルチコイドの補充療法でありストレス時にはその増量が必要とされる.塩類喪失型21-OHDの患者は9α-フルドロハイドロコルチゾンと必要に応じて塩化ナトリウムの補充が必要である.null変異のホモ接合体でホルモン補充療法によるコントロールが不良な重度の21-OHDの患者には両側副腎摘出術の適応がある.出生時から男性化を認めている女児は女性型性器形成術が必要である.思春期早発に対してはLHRHアナログによる治療がなされる.成長期にはグルココルチコイド/ミネラルコルチコイドの補充療法の適量の判断を含めた経過観察は3-4ヶ月毎に行いそれ以降は経過観察の間隔はより長期間にして差し支えない.また,精巣に関しては思春期発来後は3-5年ごとに副腎残存腫瘍をモニタリングする.
遺伝カウンセリング
21-OHDは常染色体劣性遺伝疾患である.ほとんどの親は一方が正常,もう一方が変異アレルというヘテロ接合体である.約1%の変異は新生突然変異で起こる.したがって発端者の1%は片方の親のみがヘテロ接合体である.またそれまで罹患しているとは思われていなかった親が非古典的21-OHDであると判明するケースもある.もし発端者の両親がいずれもヘテロ接合体で変異がある場合,変異アレルを両方受け継いで罹患する確率は25%,変異アレルをひとつだけ受け継いで無症候性保因者となる確率は50%,正常アレルだけを受け継いで罹患しない確率が25%である.罹患していないとわかっている同胞のうち2/3は保因者である.出生前検査が利用されており女児の男性化を軽減し,陰核形成術や膣形成術を減らすためにデキサメサゾンによる出生前治療も同時に行われることがしばしばある..
診断
臨床診断
21-水酸化酵素欠損症 先天性副腎過形成(21-OHD CAH)は以下の場合本疾患を疑う.
- 出生時に男性化を認める女児,出生後に男性化を呈する女児,
思春期早発や副腎皮質性思春期徴候を認める女児 - 小児期に男性化を認める男児(偽性思春期早発症)
- 生後4週以内に塩類喪失クリーゼを伴う新生児
検査
未治療罹患者
17-ハイドロキシプロゲステロン(17-OHP) 21-OHD患者は17-OHPの血中濃度上昇といったような生化学検査で行われる.例えば,古典型21-OHDでは20000ng/dl以上,非古典型では2000〜15000ng/dlである(図1).血漿レニン
- 血漿レニン活性(PRA)は塩類喪失型21-OHDにおいて著明な上昇を認める.また単純男性化型でも一部の患者で上昇することがある.
- レニンを直接測定することも有用である.
他の副腎ステロイド
- 血清中のΔ4-アンドロステンジオンとプロゲステロンの濃度は21-OHDの男児,女児ともに上昇する.
- 血清中のテストステロンと副腎アンドロゲン前駆体は罹患女児と思春期前の男児で上昇する.
注:塩類喪失型の21-OHD患者においては,血清中のアルドステロン濃度はPRAの上昇程度に比して不適切に低値となる.
ACTH負荷試験
17-ハイドロキシプロゲステロンとΔ4-アンドロステンジオンの濃度は基礎値および合成ACTH(コートロシン)250μgの静注後60分で測定し図1の表にプロットする.基礎値だけを測定するよりもACTH負荷試験は21-OHDの確実な診断に結びつく.その結果はCYP21A2の分子遺伝学検査によって確かめるべきである.
電解質 未治療あるいはコントロール不良の塩類喪失型の患者は血清ナトリウム濃度,クロール濃度,総CO2濃度の減少を認め,カリウム濃度の増加および尿中ナトリウム濃度の不適切な増加を認める.
核型 21-OHDの女児は46XX,男児は46XYと正常の核型を有する.保因者
正常アレルと変異アレルをひとつずつ有するものは保因者(heterozygotes)であり,無症候性である.しかし2つとも正常アレルの人に比べてACTH刺激により血清17-OHPの濃度がわずかに高値を示す(図1).ACTH刺激後の17-OHPの濃度はheterozygotesと非保因者の間でオーバーラップすることがあるためこのような検査は保因者診断のためにはもはや使用されない方法である.新生児スクリーニング
21-OHDの新生児スクリーニングには2つの目的がある.
- 生命を脅かす塩類喪失クリーゼのリスクのある古典型21-OHDの乳児を見つけること
- 外性器形態があいまいな女児の診断を円滑に進めるため
新生児スクリーニングは全てではないが非古典型21-OHDの患者も検出することができる.
21-OHDの新生児スクリーニングが義務づけられている州はNational Newborn Screening Status Report(pdf)で確認できる.17-OHPの濃度測定はほかの新生児スクリーニングで用いられているのと同じように踵より採血し,ろ紙に採取した血液スポットで行われる[Pang&Shook 1997].
- 多くのスクリーニングプログラムは単回の測定を行っており17-OHP濃度に疑問のもたれる結果に対する再検査は行っていない.
- スクリーニングの有効性を改善するために最初のスクリーニングで境界領域だった場合に再検査を行うプログラムは少ない.例えば,免疫学的検査の偽陽性率は高いため,初回の検査で陽性であった検体に対し2回目の検査として液体クロマトグラフィータンデム質量分析法による種々のホルモン(17-OHP,Δ4-androstenedione,コルチゾール)の測定を行っているプログラムもある[Minuttiら 2004].
注:(1)生後24時間以内に採取された検体では全ての新生児で値は上昇しており,偽陽性結果を示すことがある[Allenら 1997,Therrellら 1998].(2)偽陽性結果は低出生体重児[Allenら 1997]や未熟児[Saediら 1996]でも認められる.(3)本疾患の治療とは関係なくデキサメサゾンを投与された新生児においても偽陽性が認められる.
分子遺伝学的検査
遺伝子 CYP21A2が21-OHD CAHに関与している唯一の遺伝子である.
分子遺伝学的検査:臨床での利用
- 確定診断検査
- 保因者検査
- 出生前診断
- Genotype/phenotypeの治療との関係(もし塩類喪失型に関連するgenotypeであれば,ナトリウム保持性ホルモン[9α-フルドロハイドロコルチゾン]と塩分補給を行うことで塩類喪失クリーゼを防ぎ得る)
- 着床前診断
分子遺伝学的検査:臨床での応用
標的変異の分析
頻度の多い変異や遺伝子欠損を同定するパネルを用いたCYP21A2遺伝子の分子遺伝学的検査では罹患者の80-98%で原因となる変異アレルを検出できる.この変異は遺伝子変換の結果(CYP21A2が隣接する偽遺伝子CYP21A1Pに置き換わる.これには多くの有害突然変異が含まれる)として起こるか,またはCYP21A2とCYP21A1Pとの間で不等交差が起こる.[Wedell 1998].21-OHD CAHのヘテロの患者の大半は複合へテロ接合体(compound heterozygotes)である.欠失/重複の分析
変異アリルの約20%はCYP21A1P偽遺伝子の3プライム側 と隣接する補体C4B遺伝子の全体,そしてCYO21A2遺伝子の5プライム側を含んだ30Kbの欠失によって生じるキメラ偽遺伝子によって起こる(White et al 1988).サザンブロット法や1塩基置換(SNP)を利用したhomozygosity testingやmulti-ligation probe analysis (MLPA)によってこのようなlarge deletionは同定することが可能である.
シークエンス分析
遺伝子の全長シークエンシングは標的変異分析や欠失/重複分析によって変異が同定できなかった罹患患者においてまれな変異アレルを検出できることがある.
Table1 21-OHDで用いられる分子遺伝学的検査
検査法 |
同定される変異 |
変異の頻度1 |
標的変異 |
A/C659G(イントロン2スプライス変異),完全欠失やエクソン3の8塩基欠失を含む欠失変異,P30L,I172L,エクソン6のクラスター変異,V281L,F306+t,Q318X,R356W,P453S 2 |
〜80-98% |
欠失/重複 |
CYP21A2の完全欠失 |
|
シークエンス分析 |
その他のアレル |
>80-98% |
- 罹患患者の原因となるアレルの%
- 変異パネルは研究室によってさまざまである.
検査結果の解釈
シークエンス分析
シークエンス分析結果の解釈の考察に関する記載はここをクリックしてください.
標的変異分析
標的変異分析の解釈に関する記載
大規模な遺伝子組み換えでは偽遺伝子上の複数の変異が本遺伝子に生じる(Mao et al 2002).このように複数の変異が明らかになった場合はこれらの変異は同一アリル上に乗っているか,対立アリルに乗っているかを見極める必要がある.このような診断エラーを回避するには同時に両親のジェノタイプを解析するのが有効であるので可能な場合はできる限り両親の解析も同時におこなうのが望ましい.
CYP21A2遺伝子の重複も診断を誤らせる原因となる(Koppens et al 2002).保因者と判明していない人に対するスクリーニングの際に問題となる.変異のない本遺伝子と変異遺伝子を併せ持つ場合は誤って保因者と診断される可能性がある.古典的21-OHD CAH発端者の検査ストラテジー
新生児(生後初日),性器形態があいまいまたは21-OHDのリスクのある新生児は以下を必要とする.
- 完全な現病歴
- 完全な身体検査
- 骨盤内と副腎の超音波検査
- 染色体検査またはFISH法によるX,Y染色体の検出
- 血清の17-OHPと副腎アンドロゲンの濃度測定
- 17-OHPの濃度がわずか〜中等度上昇している新生児では,ACTH刺激テストで60分後の濃度を測定する.CYP21A2の分子遺伝子学的検査で21-OHD CAHの診断を確認または除外する.
- PRAと血清電解質の測定をする.症状のある罹患児,副腎クリーゼの徴候がある児ではモニタリングを続ける.
非古典的21-OHD CAH発端者の検査ストラテジー
- ACTH刺激試験の60分後
または - 早朝(8:00am前)血清17-OHP濃度測定(罹患児の基礎値は常に上昇しているわけではない)
- CYP21A2の分子遺伝子学的検査
遺伝学的に関連しているアレルの異常
CYP21A2の変異と関連している表現型はわかっていない.
CYP21A2遺伝子とTNX遺伝子を巻き込んだ隣接遺伝子症候群としてエーラース・ダンロス症候群(hypermobility type)と21-OHDの合併をきたすことがある.臨床像
自然経過
先天性副腎過形成(21-OHD CAH)は古典型としても非古典型としても現れうる(表2).
古典型21-OHDでは出生前の重要な性分化の重要な時期にテストステロンやΔ4 -アンドロステンジオンなどのアンドロジェンに曝露される結果,遺伝学的には女性である患者の外性器の男性化をきたし,時には出生時に不明瞭な外性器形態を伴う.古典型はさらに単純男性化型(〜25%)とアルドステロン合成が不十分な塩類喪失型(〜75%)に細分される.21-OHDによって塩類喪失型CAHを生じている新生児では生命にかかわる塩類喪失クリーゼをおこす危険がある.
非古典型21-OHDの患者では酵素欠損は中等度で,出生後にアンドロジェン過剰症状を示す.非古典型の女性患者では出生時の男性化はみられない.遺伝子型と臨床型との関連
これまでに明らかにされたCOL3A1遺伝子変異の約2/3では,トリプルヘリカルドメインに存在する[Gly-X-Y]343配列におけるグリシンが他のアミノ酸に置換されている.それ以外の変異の大多数はスプライス部位にありエクソンスキップを生じさせるが,より複雑な変異も存在する.mRNAの安定性を低下させる変異や蛋白の三量体形成が障害される変異など,より頻度の低い変異も報告されている.変異の型が異なっても臨床像は類似している.
COL3A1遺伝子のnull変異を有する罹患者は最初の合併症発症の時期が比較的遅いかもしれない.ただし,null変異が報告されている家系は数が少ない.
早老症を呈する表現型とIII型コラーゲントリプルへリックスのC末端の塩基配列を変化させる変異とは関係がある可能性が指摘されている.Table2 古典型,非古典型21-OHD患者の臨床像
徴候 |
21-OHD |
|
古典型 |
非古典型 |
|
出生前男性化 |
女性に見られる |
なし |
出生後男性化 |
男女とも見られる |
さまざま |
塩類喪失 |
全患者の〜 75% |
なし |
コルチゾール欠損 |
〜100% |
まれ |
古典型単純男性化型 21-OHD
副腎アンドロジェン合成過剰 子宮内での合成過剰は 46,XXの正常核型をもつ女児の出生時の男性化を招く.罹患女性ではアンドロジェン過剰によりさまざまな程度の陰核肥大,陰唇融合,尿道膣ろうの形成を招く.抗ミュラー管ホルモン(AMH)が分泌されないため,罹患女性のミュラー管から正常に子宮と卵管が形成される.罹患女児の出生時の男性化の程度からのみでは単純型21-OHDと塩類喪失型21-OHDを鑑別することはできない.
出生後,グルココルチコイド補充を受けない単純男性化型 21-OHD患者では男女とも陰毛や腋毛の早期出現,座瘡,急速な身長の伸び,骨年齢の促進といったアンドロジェン過剰による徴候が現れる.無治療の男性では陰茎の肥大と小さな精巣を認める.無治療の女性では陰核肥大,多毛,男性型頭部脱毛,月経異常,妊孕性の低下を認める.
無治療の21-OHD患児では当初の身長の伸びは急速であるが,骨端の早期閉鎖のために成人期の最終身長は低くなる.コルチゾール補充による治療を早期に開始し副腎アンドロジェン分泌をコントロールできたとしても,通常患者は期待する最終身長には到達しない.骨年齢は実年齢より促進している.思春期の発来 グルココルチコイドの治療を行われ副腎アンドロゲンの産生を抑制された児は男女ともにたいていは適切な年齢で思春期が発来する.しかし,疾患がよくコントロールされている患者の中にも例外はいる.
早期に治療を行われずにいた子供たちは,グルココルチコイドの補充療法を開始することで真の思春期早発を引き起こすことに留意が必要である.これは副腎アンドロゲンの大量分泌に由来するエストロゲンが視床下部下垂体系を抑制しておりグルココルチコイド治療を行った場合に,この抑制が解除されることで中枢性の思春期早発が起こると考えられる.妊孕性 適切に治療された女性の大部分は初潮後の月経は正常で妊娠も可能である[Loら 1999].しかし,妊娠率は低いと報告されている.その理由として,満足の得られる性交ができない不十分な膣口例やアンドロゲンの上昇により卵巣機能が低下している例,性別やセクシャルパートナーの選択に関する性心理の問題も含まれているためである.
男性 男性の生殖能低下の主な原因は精巣に副腎残存腫瘍 (adrenal rest tumor) が存在することである.それは異常な副腎組織に由来すると考えられ,グルココルチコイド療法に反応性である.さらに,下垂体性性腺機能低下症は副腎アンドロゲンや代謝産物の過剰によって下垂体のLH分泌が抑制されることによる[Ogilvieら 2006].
副腎髄質 古典的 21-OHD患者ではコルチゾールの欠乏が成長や副腎髄質機能にも影響を与え,非罹患者に比べエピネフリンやメタネフリンのレベルは低くなる[Merkeら 2000].
古典的塩類喪失型 21-OHD
21-OH機能の喪失が重度である場合,腎遠位尿細管からのナトリウム再吸収に必要な副腎アルドステロン分泌が不十分となり,患者はコルチゾール欠乏やアンドロジェン過剰とともに塩類喪失も伴う.腎性塩類喪失を伴う新生児は哺乳不良,体重減少,成長不全,嘔吐,脱水,低血圧,低ナトリウム血症,高カリウム血症を示し,副腎クリーゼ(高窒素血症,循環不全,ショック,死亡)に進展する.副腎クリーゼは早ければ出生後1から4週にもおきてくる.
外性器の形態からは認識されないため,新生児スクリーニングで発見されなかった男児は塩類喪失型副腎クリーゼの高いリスクを有する.こうした患者はしばしば診断されないまま退院し,家で塩類喪失クリーゼに苦しむことになる. 逆に性器形態があいまいな塩類喪失型の女児は早期に診断され,治療を受けることができる.非古典型 21-OHD
非古典型 21-OHDは出生後どの時期にも座瘡,陰毛の早期出現,急速な成長,骨年齢の促進,さらに古典型21-OHDと同様早期骨端閉鎖による最終的な低身長,といったアンドロジェン過剰による症状を伴って発症する.
非古典型21-OHDに罹患した女性の出生時の外性器は正常であるが,出生後に多毛,側頭部の脱毛,初経の遅れ,不規則な月経,不妊といった症状を呈してくる.21-OHDの成人女性の約60%は多毛を示すのみで,約10%が多毛と月経異常,そして約10%は月経異常のみを示す.無治療女性の妊孕性は50%と報告されている[Pang 1997].非古典型21-OHD女性の多くは多嚢胞性卵巣を生じる.
非古典型21-OHD男性は早期にあごひげが生えたり,比較的小さい精巣に大きな陰茎を伴ったりする.
非古典型 21-OHD患者における軽度のコルチゾール合成低下は臨床的な意義はない.性別と行動 古典型21-OHDの女児は出生前のアンドロゲン暴露によって外性器形態と幼少期の態度に影響を及ぼす. 出生前のアンドロゲン暴露は成人女性としての女らしさを減弱させるが,男らしさを増加させるわけではない.
幼少期の遊技行動は女性としてのジェンダーの減弱に関与し,成人してからの異性愛への興味を減弱させる.罹患女性はジェンダーに対して身体的違和感をより感じるようになるが異性愛への興味は減弱する傾向を示す.女性という性別を割り当てられることに対する満足感が少ない.逆に21-OHDの男性は幼少期の行動やジェンダーの認識,性的立場に大きな変化は見られない[Hinesら 2004]病因 21-ハイドロオキシラーゼの CYP450の機能が不十分であると,コルチゾール産生経路がブロックされ,17-OHPが蓄積される.17-OHPが過剰になるとアンドロゲンの経路へ流れ,17,20-lyaseにより17-OHPはΔ4-アンドロステンジオンに変化し,それがアンドロゲンとなる.ミネラルコルチコイドの経路は21-ハイドロオキラーゼの活性がわずかでも必要であるため,ミネラルコルチコイド欠損(塩類喪失)はこの疾患の最も重症型を呈する.
ステロイド合成の欠如は下垂体からのACTH分泌に対してネガティブフィードバック機能を損なうため慢性的なACTHによる副腎皮質刺激を引き起こし副腎過形成となる.遺伝子型と表現型の関連
アレルは残存酵素活性により重度と軽度に分けられる(表3).患者の表現型は通常,より軽症型のアレルによる酵素活性に依存する.一般に臨床的な重症度と検出される変異の間にはよい相関があるが,同じ変異を有する同一家系内の患者同士であっても臨床的重症度を常に予測できるわけではないことはしばしば経験されるがその理由は不明である.
古典的 21-OHD
遺伝子型は両アレル共に重症型の変異とされている.塩類喪失は通常酵素活性の完全喪失(in vitro解析による)による. イントロン2の終末付近でのA(またはC) からGへの点変異は古典型21-OHDで最もよく認められる変異のひとつである.イントロンの不完全なスプライシングとフレームシフトを引き起こす.この変異によるHomozygousの患者のほとんどが塩類喪失型21-OHDであるが塩類喪失の程度はさまざまである.遺伝子型と表現型が一致しないのは接合部位の変異により正常なスプライシングが行われず,互い違いにスプライシングされる部分が増加することによると説明できる.いくつかの蛋白は産生されるが,活性にばらつきが生じる[Higashiら 1988].
非古典的 21-OHD
非古典型 21-OHD患者は両アレルに軽症型の変異を有しているか,1アレルに重症型変異,1アレルに軽症型アレルを有している(compound heterozygote)と予測される.約2/3の非古典型21-OHD患者はcompound heterozygoteである.エクソン1(P30L)とエクソン7(V281L)のミスセンス変異は酵素活性を減じ,非古典的21-OHDに関与する.
Table3 残存酵素活性による,よく見られる CYP21A2 変異の分類
酵素活性 |
変異カテゴリー |
変異 |
0% |
重度(古典型) |
Null = 遺伝子欠失,大きな遺伝子転換,エクソン3の8塩基欠失,エクソン6クラスター,F306+t,Q318X,R356W |
<1% |
A/C659G (イントロン2スプライス部位変異)(I2G) |
|
2-11% |
I172N |
|
〜20-50% |
軽度(非古典型) |
P30L,V281L,P453S |
専門用語について
先天性副腎過形成(一般的にCAHと省略される)は特異酵素欠損(例えば,21-ハイドロオキシラーゼ欠損)を前置きとして記載する傾向にあり,それが望ましい.
21-OHD CAHには以前は以下のものが含まれていた:副腎性器症候群(AG症候群),C-21-ハイドロオキシラーゼ欠損症,先天性副腎皮質過形成
非古典型21-OHD CAHは以前は軽度のあるいは遅発性の病型として言及されていた.
塩類喪失型21-OHD CAHはsalt-wasting CAHまたはsalt-losingCAHとよばれる.頻度
古典型21-OHD CAH 約650万人の異なる人種における新生児スクリーニングのデータ解析によれば古典型21-OHDの頻度は出生15,000人あたり1人である.
人種ごとの頻度
- アラスカのYupikエスキモーでは1/300
- サウジアラビアでは1/5,000
- ヨーロッパと北米が1/10,000-16,000
- 日本人では1/21,000
- ニュージーランドでは1/23,000
非古典型21-OHD CAH 多彩な人種で構成されるニューヨーク市における非古典型の頻度は1/100と推定されている.非古典型21-OHDの頻度が最も高い人種はアシュケナジーユダヤ人で1/27である.その他頻度の高い人種集団はヒスパニック(1/40),スラブ人(1/50),イタリア人(1/300)である.
鑑別診断
本稿で扱われる疾患に対する遺伝学的検査の実施可能性に関する最新情報は,GeneTests Laboratory Directoryを参照のこと.―編集者注.
副腎皮質の束状層におけるコルチゾール生合成には 5つの酵素による合成ステップをふむ.先天性副腎過形成(CAH)はこのいずれの酵素の欠損によっても生じる.コルチゾールの合成障害と分泌低下はACTHの慢性的な上昇を招き,これが副腎の過剰に刺激して過形成をきたす.CAHの5つの型をTable4に示す.副腎皮質の生合成のそれぞれのステップにおける酵素機能障害が前駆体を蓄積させ欠損物質が生じる.21-OHDはCAHの90%以上を占め,最も多い.
Table4 CAHをきたす酵素欠損
|
- 男性では出生時男性化はない.
- 塩類喪失を伴う.
- 高血圧を伴う.
- 女性では出生時もしくはその後男性化を示す.
非古典型 21-OHD CAHアンドロジェン過剰による症状を示す女性では非古典型 21-OHDを考慮する必要がある.アンドロジェン過剰を伴う女性における非古典型21-OHDの頻度は一般人口の1-3%と報告されているが,人口集団によってはそのような女性の頻度はもっと高い.
チトクロームP450酸化還元酵素欠損症 CAHのまれな病型でTable4には含まれていない.CYP450酸化還元酵素欠損症でPORの変異による.P450C17(17-ハイドロキシラーゼ)およびP450C21(21-ハイドロキシラーゼ)の2つのステロイド合成酵素の部分欠損が同時に存在することが尿中ステロイド排泄から示唆される.注目すべきは,CYP450酸化還元酵素がNADPHからの両酵素への電子伝達に重要な役割を持っているということである.
CYP450酸化還元酵素欠損症の表現型は単一のステロイド異常症から古典型Antley-Bixler症候群(ABS)まで幅広い.POR欠損者はコルチゾールが欠如しており臨床的にたいしたことのないものから生命の危機に及ぶものまである.新生児男児は外性器形態があいまいで,陰茎は小さく停留精巣を認める.新生児女児は膣閉鎖症を認め,小陰唇の融合,大陰唇の低形成,陰核肥大を呈する.ABSの頭蓋顔面の特徴は,POR領域での最も重症例では頭蓋骨癒合症,後鼻孔狭窄または閉鎖症,外耳道狭窄,水頭症を起こすことがある.骨奇形では橈骨上腕骨癒合,新生児期骨折,先天性長幹骨湾曲,屈指症,関節性拘縮,クモ指症,内反足を起こすことがある.
遺伝形式は常染色体劣性遺伝である.臨床的マネジメント
病気の程度を確定するための初期診断における評価
塩類喪失型を評価するために:
- 血清レニン活性(PRA)またはレニン濃度
- 血清電解質
21-OHD CAHの古典型と非古典型を区別するために:
- 17-OHP,Δ4-アンドロステンジオン,コルチゾール,アルドステロンの基礎値
- ACTH刺激試験を実施し,基礎値と刺激後の17-OHPの濃度の比較
女児において出生前の男性化の程度評価のために:
- 外性器形態とその開口についての身体所見の注意深い観察
- 尿道と膣の解剖学的評価のため膣造影
男児,女児ともに出生後の男性化の程度評価のために:
- 骨年齢評価
- 血清の副腎アンドロゲン濃度(DHEA,Δ4-アンドロステンジオン,テストステロン)
病気の徴候に対する治療
臨床医は早期に治療を開始し,もしあるのならばコルチゾール欠乏や鉱質コルチコイド欠乏による影響をとどめるために, 21-OHDの診断をできる限り早期につける義務がある.
小児内分泌,小児泌尿器,臨床遺伝学,精神科などのさまざまな専門分野のチームがあいまいな外性器の児の診断や治療に必要となる[Hughesら 2006].古典型21-OHD CAH
グルココルチコイド補充療法 グルココルチコイド補充療法の目標は,欠損しているステロイドの補充をおこない,副腎性ホルモンとグルココルチコイド過剰分泌をできる限り少なくすることであり,男性化を予防し成長の可能性を最大限に伸ばし,妊孕性を促進することである[Claytonら 2002].
CAHの治 療では基本的にグルココルチコイド補充が必要となり,通常 1日2ないし 3回に分けてハイドロコーチゾン (24 時間あたり 10-20 mg/m 2)が経口的に投与される.小児におけるグルココルチコイド療法は,医原性クッシング症候群を起こさないように,そして正常な成長および骨成熟がおこるように副腎アンドロジェン分泌の抑制をするというバランスが求められる.
過剰な治療はクッシング様徴候を招くので避けなければならないが,しばしば血清17-OHP濃度が生理学的範囲に低下した場合におきてくる.したがって,治療中の患者における17-OHPの正常範囲は,アンドロジェンが性別や性的発達段階に応じた適切なレベルに維持されているという条件で,通常よりも高めに設定(100-1,000 ng/dl)すべきである.
ストレス状態下(手術,発熱性疾患,ショック)ではすべての 21-OHP患者はより多量のグルココルチコイドを必要とする.典型的には通常の2-3倍量が経口的または経静脈的に投与される.
罹患患者は緊急時のステロイド投与量に関する医療情報を携帯するべきである.
古典型21-OHD CAHの患者は生涯にわたってグルココルチコイド投与が必要となる.成長が完了した後はより力価の高いグルココルチコイド(プレドニゾロン,デキサメサゾンのような)を用いることができる.これらは小児では成長を抑制する傾向がある.
鉱質コルチコイド補充療法 塩類喪失型の 21-OHD患者では,9 α-フルドロヒドロコルチゾン(フロリネフ)(0.05-0.3 mg/日 経口)と塩化ナトリウム(3 g/日を処方や食事に加える)が必要となる.
塩化ナトリウムの補充は幼少期以降は必要としないかもしれない.同様に一日に必要とされる鉱質コルチコイドの量も年齢とともに減少してくる.副腎摘出術 null変異のホモ接合体でホルモン補充療法では良好なコントロールが得られなかった重度21-OHD患者に対する両側副腎摘出術が報告されている[Van Wykら 1996,Meyers&Gru 2000].こうした患者はアジソン病患者に対する治療のように,より適切に治療できるかもしれない.
副腎摘出術を受けた21-OHD患者の女性で術後に副腎ステロイドホルモン濃度が再上昇した例があることを心にとどめておく必要がある.この再上昇は卵巣に異所性副腎皮質組織が存在していたためと考えられる.これら患者の意味と遺残組織の頻度を明らかにするにはより長期のデータが必要である.女性型の性器形成術 生下時に男性化を伴っている古典型 21-OHD女性では,不要な勃起組織を除去し,性的に敏感な陰核を保存し,月経,性交,分娩に適切な正常の膣開口部を増設する女性型性器形成が行われる.陰核の手術は典型的な場合早期(できれば6-18か月)に行われる.膣形成術が必要な場合は,手術は思春期後期に行われる.これは膣開口を保つために膣拡張が必要とされるためである.
思春期早発 21-OHD CAHにおこる思春期早発は黄体ホルモン放出ホルモン (LHRH)誘導体による治療が行われる.
思春期から大人への移行 21-OHD CAH に対する治療の改良によって良好な予後および正常と変わらない平均寿命が得られる.成人における治療の目標は,幼少期の主な目標であった正常な成長の維持から妊孕性の維持,健全な性機能,骨塩量などを含めた全身的な健康状態の維持,心血管系疾患のリスクマネージメントへと移行する.CAHの成人患者は精神的サポートなどを含め多くの専門分野によるアプローチが必要となる[Ogilvieら 2006].実際,成人に対するエビデンスに基づく治療プロトコールが不足している[Kruseら 2004].非古典型21-OHD CAH
非古典型21-OHD患者は必ずしも治療を必要としない.多くは生涯にわたって無症状であり,あるいは症状が思春期,思春期後,あるいは閉経後に現れる.伝統的に古典型21-OHD患者に対するよりも低用量のグルココルチコイドによる治療が行われてきた.治療の適応となる症状は骨年齢の促進,重度の座瘡[Degitzら 2003],多毛,月経不順,不妊が問題になる場合である.初期症状の予防
塩類喪失クリーゼ 新生児スクリーニングプログラムは古典型21-OHDの新生児を同定し,生命を脅かす塩類喪失クリーゼの可能性がある児に対して事前に初期治療を行う目的がある.
性器形態があいまいな女性 胎児DNAの分子遺伝子学的検査は,21-OHD CAHにおける出生前診断が可能である.女性の胎児で妊娠初期から出産まで母親がデキサメサゾンを投与されることで,胎児期のアンドロゲン産生を抑制でき性器形態のあいまいさを軽減またはなくすことができる.
症状に対する治療 グルココルチコイド補充療法,ミネラルコルチコイド補充療法二次的合併症の予防
低身長 低身長はグルココルチコイドの過剰治療による成長抑制あるいは不十分なグルココルチコイド治療による骨成熟の促進によっておこる.単独あるいはゴナドトロピン放出ホルモン (GnRH )と併用したヒト成長ホルモン注射は重度の成長障害[Quintosら 2001]を伴う21-OHD患者の身長を伸ばすのに用いられる[Lin-suら 2005].定期検査
以下の評価は子供たちが活発に成長するにあたり3-4カ月ごとに行われるべきである.それ以降はより間隔をあけて行われる.評価の頻度は個々の患者の必要性に応じて変更するべきである.グルココルチコイド補充療法の効果は以下の測定でモニターする:
- 早朝血清中の17-OHP,Δ4-アンドロステンジオン,テストステロンの濃度を新生児期はだいたい3ヶ月毎に,その後は3-6ヶ月毎に測定.(いくつかの例では,24時間尿中サンプルを用いて尿中のプレグナントリオール,17-KSがホルモンコントロールの評価に役立つことがある.しかし,蓄尿法は単純採血に比べて実用的ではない.)
- 身長の伸び,体重増加,思春期の発達,コルチゾールやアンドロゲン過剰の臨床徴候.
- 骨成熟度を評価するために骨年齢(6-12カ月間隔で)
ミネラルコルチコイド補充療法の効果は以下の測定でモニターする:
- 血圧
- 早朝血清レニン活性または直接レニン分析(たいていは直立姿勢で)
男性における精巣異常のモニタリング 超音波やMRIを用いて精巣の定期的な画像診断を思春期以降より開始,3-5年ごとに繰り返し行われるべきである.
リスクのある血縁者の検査
もし21-OHD CAHの出生前診断が行われていなかった場合,早期診断と治療を容易にするために潜在的に危険な状態にある血縁者の新生児スクリーニングの血液サンプルより17-OHPを測定するほうがよい.研究中の治療
政府による臨床トライアル 疾患の広がりや状態に関し臨床研究情報の評価を行う政府の臨床研究調査.注:この疾患に対する臨床トライアルはない
その他
古典型 21-OHD患者の妊娠 古典的塩類喪失型 21-OHDの妊婦は綿密に内分泌専門医のモニターを受ける必要がある.妊娠中には副腎アンドロジェンが増加する傾向にあるので,グルココルチコイドやミネラロコルチコイドの必要量は通常増加する.母体からの過剰な副腎アンドロジェン産生にもかかわらず,女性胎児の外性器は必ずしも男性化を示さない[Loら 1999].遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
常染色体劣性遺伝である.
患者家族のリスク
発端者の親
- ほとんどの両親はヘテロ接合体で,正常アレルと変異アレルを有している.
- ヘテロ接合体は ACTH負荷を行ったときの17-OHPレベルが正常アレルのみを持つ人に比べてわずかに上昇していることがあるものの,無症状である.
- 約1%の変異は新生突然変異で生じるので,発端者の1%では両親のうち一人だけがヘテロ接合体である[Kroneら2000].
- 時に罹患しているとは思われていなかった親が非古典型 21-OHDであることが判明する.両親が非古典型21-OHDを有していないかどうか明らかにするために,分子遺伝学的検査やホルモン検査を施行するのは適切である.
発端者の同胞
- もし患者の両親がヘテロ接合体であるならば,個々の同胞について,変異アレルを 2つ受け継ぎ,その結果罹患する確率は25%,変異アレルを1つだけ受け継いだ無症候保因者である確率が50%,正常アレルのみを受け継いだ非保因者である確率が25%である.
- 発症していない同胞が保因者である確率は 2/3である.
- もし発端者の一方の親がヘテロ接合体でもう一方の親が 21-OHDに罹患している場合,個々の同胞は50%の確率で変異アレルを2つ受け継いで罹患し,50%の確率で変異アレルを1つ受け継いで保因者となる.
発端者の子 罹患者の子は50%のリスクでCOL3A1遺伝子変異を受け継ぎ,発症する.
- 罹患した患者は子供に変異アレルを 1つ伝える.
- 21-OHDの保因者頻度が高いので,患者のパートナーに対して CYP21A1 の分子遺伝学的検査を提供するのは適切である.
- もしパートナーが非保因者であれば,子が21-OHDに罹患する可能性はずっと低くなる.(変異解析は変異を100%検出できるわけではないので,遺伝子の全領域をシークエンスしない限り,パートナーが保因者である若干の可能性は残される.)
- もしパートナーが既知の変異の保因者であった場合,二人の子が罹患する可能性は50%である.遺伝子型にもとづいて臨床型を予測するのは不完全にしかできない.
発端者の他の家族
発端者に変異を伝えた親の同胞も 50 %の確率で保因者である.保因者の検出
分子遺伝子学的検査 CYP21A2 遺伝子の分子遺伝学的検査による保因者検査は,発端者に原因となる変異が同定された場合にリスクのある親族に対して提供可能となる.
ホルモン検査 保因者では非保因者に比べて ACTH負荷時の17-OHPレベルがわずかに高くなるが,これは保因者と非保因者との間でオーバーラップがある.したがって,保因者検査には分子遺伝学的検査がより望まれる.遺伝カウンセリングに関連した問題
遺伝子型と表現型の関連
21-OHD CAHのほとんどの患者において遺伝子型が疾患の重症度を予測するのに有用である.常染色体劣性遺伝であるため,表現される表現型は患者のアレルの変異のより軽症型の方が反映される.それぞれの変異の重症度はin vtroにおける発現実験による残存酵素活性のパーセンテージで分類される.表3に頻度の高い変異と表現型が記載されている.塩類喪失型の患者は通常,もっとも重度の変異を有しており(欠失のホモ接合性),一方,非古典型21-OHD CAHの患者はより軽度のV281L変異[Wilsonら 1995]などを有する.しかし,中等度の変異のグループ内ではかなりの相違が見受けられる[Kroneら 2000].頻度が多いわけではないが,遺伝子型と表現型の不一致はV281L,P30L,I172Nのようなそれほど重度ではないアレルの変異によっておこる.さらに,イントロン2のスプライス変異はたいてい塩類喪失の重症度の表現型バリエーションに関与している.出生前診断の背景として,出生前治療の必要性を決定するために古典型と非古典型を区別することが重要である.まれな例外:V281LやP30L変異を有する罹患患者の少数(<3%)においてはその変異は非古典型の表現型となることが期待されたが古典型の表現型を呈した.;Stikkelbroeckら(2003)はI172Nともうひとつ重度の変異をもっている患者では古典型が予想されるが,少数%のみが古典型であり,ほとんどが非古典型の表現型を呈したことを報告した.
発端者である患者が男性化を呈する女児のケースでは,次女児の男性化の程度の予想が可能である.発端者が男児の場合は遺伝子型に基づいた次女児の表現型の予測は不可能であり,外性器の男性化の軽減を図るために出生前治療を行うべきである.家族計画 遺伝学的リスク,保因者に関する問題,出生前診断と治療に関しては妊娠前に情報提供すべきである.
DNAバンク DNAバンクとは将来的な使用を想定してDNAを(多くは白血球から抽出する)保存しておくものである.遺伝子検査技術や遺伝子,変異,疾患に対するわれわれの認識が将来変化するかもしれないので,現在の遺伝子検査の精度がすべての変異を検出できないような場合にはDNA保存が考慮されうる.出生前診断
高リスク妊娠 古典型 21-OHDのリスクがある妊娠に対する出生前診断は数十年前から行われており,罹患女性の男性化を軽減し陰核形成術,膣形成術の必要性を減らすことができるデキサメサゾンによる出生前治療は,1984年以降行われて成功を収めている.出生前治療のプログラムには以下の項目が含まれるべきである(図2)
- 妊娠前の遺伝子カウンセリング
- 妊娠前の発端者と両親の分子遺伝子学的検査.疾患を引き起こす原因であるCYP21A2変異と両親が保因者であることを確かめるためにおこなう.
- 出生前診断が未施行の9週以前の妊娠が確認されたら,胎児の過剰な副腎アンドロジェン産生を抑制し,罹患女児の男性化を軽減するために,妊婦に対してデキサメサゾン(20 μg/kg/日)を1日3回に分けて投与する.
- 胎生 10-12週の時期に絨毛採取もしくは15-18週の羊水穿刺によって胎児細胞を採取し,細胞遺伝学的方法かY染色体特異的プローブを用いて胎児の性別を判定する.(治療法の決定は出生前診断の結果に基づくため,妊娠のなるべく早い時期に結果を得るほうが有利である.したがって胎生10-12週の絨毛採取のほうが望まれる.)
- もし胎児が女性で,発端者で CYP21A2 遺伝子の両アレルの変異がすでに確認されているのであれば,胎児の両アレルに変異があるかどうかの分子遺伝学的検査を行う.
- もし胎児が男性であったり(細胞遺伝学的検査や Y染色体特異的プローブによる)非罹患女児であったり(DNA検査による)した場合には,デキサメサゾンによる治療は中止する.
- もし胎児が女性で DNA検査で21-OHD罹患が判明したり,あるいは判定ができない状況である時はデキサメサゾン治療を分娩時まで続ける.
注:(1)出生前治療は,申請時期あるいはそれ以降のホルモン補充療法の必要性には影響はない.(2)一般的に,非古典型21-OHDのリスクがある胎児への出生前治療は推奨されていない[Clayton 2002].(3)羊水穿刺による17-OHP濃度測定や連鎖解析によるHLAタイピングを用いた出生前診断は分子遺伝学的技術の進歩にともない CYP21A2 の解析にとってかわられた.
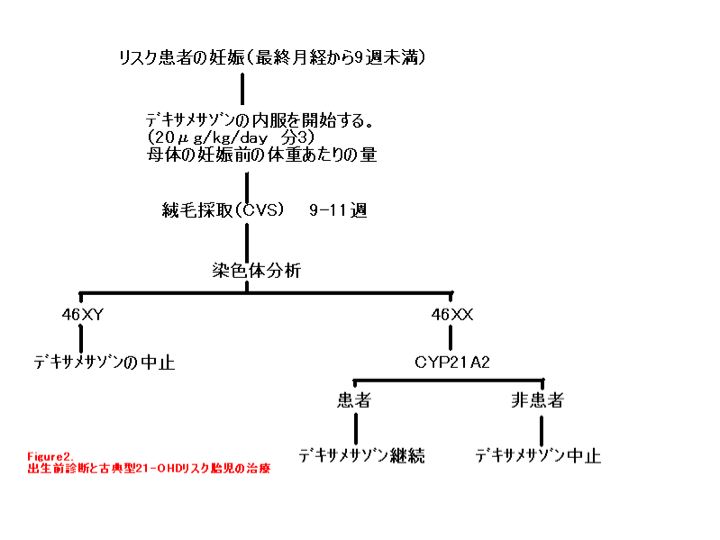
注:胎生週数は最終月経の開始日あるいは超音波検査による測定に基づいて計算される.
適切な投与が行われれば,デキサメサゾンは罹患女児の外性器の男性化を防ぐのに有効である.デキサメサゾンによる出生前治療は妊婦と胎児の双方にとって十分許容できる[Newら 2003]が,デキサメサゾン投与を受けた母では統計学的に有意に体重増加に伴う皮膚線条や浮腫が増加するという報告もある.これまでで最大の調査では,患者と医師が推奨された治療プロトコールを忠実に行ったという条件では,胎児の先天異常の増加はないし,出生時体重,出生時身長や頭囲にも対照と比較して差は認められていない[Forestら 1989,Lajicら 1998,Newら 2001] .さらに,Forest, Lajic ,Trautman による経過追跡では出生前治療を受けた子どもたちの成長と発達は正常である[Trautmanら 1995,Forest 1998, Lajic ら 1998].しかしながら,妊娠中一部の期間もしくは全期間デキサメサゾン治療を受けた子の長期経過観察は今後も続けていく必要がある.
低リスク妊娠 出生前の超音波検査の使用頻度の増加や解像度の改善によって胎児期の生殖器異常や副腎異常がより高頻度に同定されるようになっている[Saadaら 2004].Pinhas-Hamielらは2002年に,出生前に超音波検査をされた10000人の胎児のうち16人で性器形態異常を同定した.16人中3人は最終的に21-OHD CAHの診断であった.
もし定期超音波検査で外性器形態があいまいであった場合には,胎児の染色体型,SRY遺伝子に対するFISH,そしてミューラー管構造物の超音波による評価が必要である.46XX,SRY(-),子宮形態が正常の胎児は古典型21-OHDを考慮しなくてはいけない.羊水穿刺にてCYP21A2の分子遺伝子学検査が望ましい.
低リスク妊娠での罹患胎児の診断は出生前治療をするためにするわけではないが,21-OHDの出生前診断は新生児期のマネージメントや家族の準備の面で意味のあることである.
着床前遺伝子診断(PGD)は疾患の原因となる変異が以前に同定されているメンバーのいる家族にとっては有用かもしれない.分子遺伝学
分子遺伝学の情報は教科書に記載されているものと違うであろう.より最近の情報が含まれている.21-OHD CAHの分子遺伝学
遺伝子表記 |
染色体の座位 |
蛋白名 |
CYP21A2 |
6p21.3 |
チトクロームP450 21 |
データは以下の参考文献より集められている
:遺伝子の表記はHUGOより,染色体の座位はOMIMより,蛋白名はSwiss-Prot.より.
21-OHD CAHに関するOMIMの記載
201910 副腎過形成,先天性,21水酸化酵素欠損症のため |
21-OHD CAHに関するゲノムデータベース
Gene Symbol |
Entrez Gene |
HGMD |
GeneCards |
GDB |
GenAtlas |
CYP21A2 |
201910 |
CYP21A2 |
CYP21A2 |
120605 |
CYP21A2 |
正常アレルのvariant:副腎の21水酸化酵素の機能性遺伝子,CYP21A2は非機能性の偽遺伝子CYP21A2Pから30kbほどの場所にありHLA遺伝子クラスターの6p染色体上に存在する.
CYP21A2とCYP21A2P遺伝子は10個のエクソンを有する.CYP21A2遺伝子は5つの正常のバリエーションがある(655C>A, insL9, K102R, D183E, S268T, N493S).病因アレルのvariant:CYP21A2とCYP21A2Pのあいだには類似シークエンスが多く(96-98%),どうやら組み替え現象が可能なようである.(1)CYP21A2の欠失・重複が補足的に起こる結果,減数分裂の間に不等交差が起こる.(2) 変異を不活化するようなCYP21A2とCYP21A2Pの間の変換現象がCYP21A2P からCYP21A2へ転写させる.CYP21A2P からCYP21A2へ転写させる際に非機能性偽遺伝子において9個の原因となる変異が機能性遺伝子を不活化させる.これらの9つの変異はCYP21A2欠失と大きな遺伝子変換とともに原因となるCYP21アレルの95%ほどの割合を占める.
100以上の変異がある.点突然変異,小さな欠失,小さな挿入,遺伝子の複雑な再配列が含まれる.正常遺伝子産物:コードされている蛋白は494のアミノ酸からなり分子量55kdの蛋白である.この酵素は多くとも28%のその他のチトクロームP450酵素と同種である.
異常遺伝子産物:異常産物は特異的変異に依存する.およそ20%の変異は30Kbの欠損である.イントロン2のスプライス変異は20−30%の頻度で認められスプライス異常により短縮した不完全なタンパクを生じる(Higashi et al 1988).
更新履歴
- GeneReview 最終更新日: 2002.2.26. 日本語訳最終更新日: 2003.8.31.
日本語訳者: 櫻井晃洋(信州大学医学部附属病院遺伝子診療部)
- Gene Review 最終更新日: 2006.9.7. 本語訳最終更新日: 2007.9.21
日本語訳者: 塚本幸子,臼井健(国立病院機構京都医療センター)