CASK異常症
(CASK Disorders)
[Synonyms: Broad Thumbs-Hallux Syndrome]
GeneReviews著者: Ute Moog, MD and Kerstin Kutsche, PhD
日本語訳者:西村直人(防衛医科大学校病院 小児科)、 黒澤健司(神奈川県立こども医療センター 遺伝科)
GeneReviews最終更新日: 2020.5.21. 日本語訳最終更新日: 2021.5.21.
原文: CASK Disorders
要約
疾患の特徴
CASK異常症は、女性と男性の両性で表現型スペクトラムを有する。主に2つのタイプの臨床所見が認められる。
- 一般的にCASK遺伝子の機能喪失変異に関連している橋小脳低形成を伴う小頭症(MICPCH)
- 一般的にCASK遺伝子のhypomorphic病原性変異に関連している眼振を伴う、または伴わないX連鎖性知的障害(XLID)
MICPCHは通常、中等度から重度の知的障害、眼科的異常を伴う、または伴わない 進行性の小頭症、および感音性難聴を有する女性に認められる。多くの場合、独座は可能で、20%~25%は歩行が可能である。多くの場合、言語はほとんど獲得されない。神経学的特徴には、体軸性の筋緊張低下、四肢の過緊張/痙縮、ジストニアまたはその他の運動障害が含まれる。10歳までに40%近くがてんかん発作を起こす。行動には、睡眠障害、手の常同運動、および自傷行為がある。
男性のMICPCHは、重度から最重度の発達遅滞に加えて、重度のてんかん性脳症を伴う場合と伴わない場合がある。てんかん発作がある場合は、早期に発症し難治性の場合もある。
軽度の(すなわち、hypomorphic)病原性変異を有する患者および家族では、臨床的表現型は通常、眼振および他の臨床的特徴の有無にかかわらず、XLIDである。男性は、眼振および他の眼症状の有無にかかわらず、軽度から重度の知的障害がある。女性は通常、正常な知能を有し、一部の患者は眼症状を伴う、または伴わず、軽度から重度の知的障害を呈する。
診断・検査
CASK異常症は、分子遺伝学的検査で、女性ではCASK遺伝子のヘテロ接合性病原性変異、男性ではCASK遺伝子のヘミ接合性病原性変異を同定することで確定診断となる。まれに、罹患男性がモザイクの病原性変異を有することがある。
臨床的マネジメント
症状の治療:
治療は、対症療法的であり、発達遅滞と知的障害の問題に対する標準的な管理、てんかん発作に対する薬剤、栄養のサポート、理学療法の使用、視覚障害または難聴の治療などを含む。
遺伝カウンセリング
CASK異常症はX連鎖性遺伝形式である。CASK異常症の発端者の家族へのリスクは、発端者の表現型(すなわち、MICPCHまたはXLID±眼振)に依存する。
- MICPCH
ほとんどの罹患女性および罹患男性は、孤発例(すなわち、1家系に1人だけ発症) であり、CASK遺伝子の新生病原性変異の結果としてこの疾患を発症する。ヘテロ接合性の女性はこの表現型を呈するため、無症状の母親がCASK遺伝子のヘテロ接合性病原性変異を有する可能性は低い。発端者が孤発例の場合、同胞への再発リスクは低いが、親の生殖細胞モザイクの可能性があるため、一般集団よりもリスクは高くなる。
- XLID±眼振
CASK異常症男性の父親は、この疾患ではなく、CASK遺伝子のヘミ接合性病原性変異も有さない。男性が唯一の罹患家族の場合、母親がヘテロ接合性であるか、または罹患男性が新生病原性変異を有する可能性がある。複数の罹患者を有する家族においては、罹患男性の母親は絶対的ヘテロ接合体である。発端者の母親がCASK遺伝子の病原性変異を有している場合、各妊娠でその変異を受け渡す確率は50%である:病原性変異を受け継いだ男性は発症する。病原性変異を受け継いだ女性は、一般的には無症状だが、様々な症状を呈する可能性がある。CASK遺伝子の病原性変異が母体の白血球DNAから検出できない場合、親の生殖細胞モザイクの可能性があるため、同胞へのリスクは一般集団よりは高くなる。
CASK遺伝子の病原性変異が罹患した家族内で確認されている場合、リスクのある妊娠のための出生前検査や、CASK異常症の着床前遺伝学的検査を検討することがある。
GeneReviewの記載範囲
| CASK異常症:含まれる表現型1 |
|---|
|
同義語と旧式の名称は命名法を参照すること。
- これらの表現型の他の遺伝的要因については,鑑別診断を参照のこと。
診断
CASK異常症は、眼振伴う、または伴わず、軽度~重度の知的障害から、中等度~最重度の知的障害およびMICPCHまで、幅広い表現型スペクトラムに関連しており、しばしばてんかん発作を伴う。CASK異常症はX連鎖性疾患であり、男性よりも女性に多く報告されている。女性のMICPCHは、現在最も一般的な表現型である。
示唆的な所見
CASK異常症は、知的障害および以下の追加的な所見のいずれかを有する患者において考慮されるべきである。
・-10SDまでの進行性小頭症
- 橋および小脳低形成
- 筋緊張低下、筋緊張亢進、またはその両方の組み合わせ(中枢性の筋緊張低下と四肢の筋緊張亢進)
- てんかん発作(大田原症候群、ウエスト症候群又はミオクロニーてんかんからなる早期及び難治性てんかん発作を含む)
- 眼振、斜視、視神経低形成、および/または網膜異常症
- 感音性難聴
- 低身長
CASK異常症の診断は、CASK遺伝子のヘテロ接合性病原性変異の女性と、CASK遺伝子のヘミ接合性病原性変異の男性で確立される(表1参照)。
注:まれに、罹患男性がモザイクの病原性変異を有することがある。
CASK異常症の表現型は、しばしば知的障害、小頭症、および/または脳小脳の低形成を伴う他の多くの遺伝性疾患と区別がつかないため、推奨される分子遺伝学的検査のアプローチには、マルチ遺伝子パネルまたは網羅的ゲノム検査が含まれる。
注:単一遺伝子検査(CASK遺伝子のシークエンス解析後、標的遺伝子の欠失/重複解析を行う)が有用なことは稀であり、通常は推奨されない。
- CASK遺伝子やその他の関連のある遺伝子(鑑別診断を参照)を含む知的障害や脳構造異常、または橋小脳の低形成に特化したマルチ遺伝子パネルは、最も合理的なコストにより、症状の遺伝学的原因を特定できる可能性が高く、一方で、意義不明の変異や基本的な表現型を説明できない病原性変異が検出されることを最小限にすることができる。
注:(1)パネルに含まれる遺伝子および各遺伝子に使用される検査の診断感度は検査室によって異なり、時間経過とともに変化する可能性がある。(2)マルチ遺伝子パネルの一部には、このGeneReviewで議論されている疾患とは関連性のない遺伝子を含んでいるものもある。(3)一部の検査室では、パネルのオプションとして、検査室が設計したカスタムパネル、あるいは臨床医が指定した遺伝子を含む表現型に特化したカスタムエクソーム解析が含まれる場合がある。(4)パネルで使用される解析方法には、シークエンス解析、欠失/重複解析、および/またはその他のシークエンスに基づかない検査が含まれる。
この疾患については、欠失/重複解析を含むマルチ遺伝子パネルが推奨される(表1参照)。
マルチ遺伝子パネルの概要については、ここをクリックする。遺伝子検査を依頼する臨床医のためのより詳細な情報は、ここを参照のこと。
- 網羅的ゲノム検査(どの遺伝子が関与している可能性が高いかを臨床医が判断する必要がない)も良い選択肢である。エクソームシークエンシングが最も一般的に用いられているが、ゲノムシークエンシングも可能である。
使用するシークエンシング法によっては、シングルエクソン、マルチエクソン、または全遺伝子欠失/重複が検出されない場合がある。使用したシークエンシング法によって変異が検出されない場合は、次のステップとして、標的遺伝子欠失/重複解析(エクソンおよび全遺伝子欠失/重複を検出するためのエクソームアレイ解析またはマイクロアレイ染色体検査を含む場合がある)を行う。
網羅的ゲノム検査の概要については、ここをクリックする。遺伝子検査を依頼する臨床医のためのより詳細な情報は、ここを参照のこと。
注:(1)少数の男性において、ヘミ接合体でのCASK遺伝子再構成や、モザイクでのCASK遺伝子再構成や欠失挿入変異が報告されている[Saitsu et al 2012, Moog et al 2015, Hayashi et al 2017]。(2)シークエンス解析や欠失/重複解析で病原性変異が同定できず、それでもCASK異常症の疑いが高い場合には、核型解析が適切かもしれない。CASK遺伝子を障害する均衡型Xp逆位を有する2例の女性が報告されている[Najm et al 2008;K Kutsche, unpublished]。
表 1. CASK異常症に用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | その方法により検出可能な病原性変異を有する発端者の割合2 |
|---|---|---|
| CASK | シークエンス解析3, 4 | ~70%5,6 |
| 標的遺伝子の欠失/重複解析7 | ~30%5, 6, 7 | |
| マイクロアレイ染色体検査8 | ~28%5, 8 | |
| 染色体検査 | 稀9 |
- 染色体座位とタンパク質については、表A.遺伝子とデータベースを参照。
- この遺伝子で検出されたアレル変異の情報については、分子遺伝学を参照。
- シークエンス解析は、良性変異、良性の可能性が高い変異、意義不明な変異、病原性の可能性がある変異、病原性変異を検出する。病原性変異には、小さな遺伝子内欠失/挿入とミスセンス変異、ナンセンス変異、スプライス部位の変異が含まれる場合がある。典型的には、エクソンまたは全遺伝子の欠失/重複は検出されない。シークエンス解析結果の解釈で考慮すべき問題については、ここをクリック。
- シークエンス解析前のPCRによる遺伝子増幅の欠如は、罹患男性におけるX 染色体上の(マルチ)エクソンまたは全遺伝子の欠失を示唆する。確定には標的遺伝子の欠失/重複解析による追加検査が必要となる。
- データは、Human Gene Mutation Databaseより引用している[Stenson et al 2017]。
- 割合は女性の発端者に基づいている。生存している女性の発端者は、シークエンス解析によって検出された変異を有する可能性が高い(遺伝子型-遺伝子型の相関を参照)。
- 標的遺伝子の欠失/重複解析は、遺伝子内の欠失または重複を検出する。定量PCR、long-range PCR、MLPA法、単一エクソン欠失や重複を検出するために設計された標的遺伝子マイクロアレイ染色体検査などの方法が用いられる。標的遺伝子の欠失/重複検査は、単一エクソンから全遺伝子欠失まで検出するが、大規模な欠失や隣接遺伝子の欠失のブレークポイント(Moog et al [2011]、Burglen et al [2012]、Hayashi et al [2012]、Hayashi et al [2017])は、検出できない場合がある。
- マイクロアレイ染色体検査(CMA)は、オリゴヌクレオチドまたはSNPアレイを用いて、シークエンス解析では検出できないゲノム全体の大規模な欠失/重複(CASK遺伝子を含む)を検出する。報告されているほとんどのCASK遺伝子の欠失/重複は、CMAによって検出できるほどの大きさである。欠失/重複の大きさを決定できるかどうかは、使用するマイクロアレイの種類とXp11.4領域のプローブの密度に依存する。現在臨床で使用されているCMAデザインはXp11.4領域を標的としている。
- CASK遺伝子を障害する均衡型Xp逆位を有する2人の女性が報告されている[Najm et al 2008; K Kutsche, unpublished]。
臨床的特徴
臨床像
CASK異常症は女性に多く報告されており、女性と男性で異なる表現型スペクトラムが含まれている。
- 女性は通常、中等度から重度の知的障害を有し、ほとんどが進行性のMICPCHを有する。考えられる所見としては、眼科的異常および感音性難聴がある。XLID±眼振の表現型を有する男性の血縁者である女性は、まれに軽度から重度の知的障害の表現型を示すことがある。
- 男性では、重度(知的障害およびMICPCH、または早期乳児てんかん性脳症[大田原症候群、ウエスト症候群、または早期ミオクロニーてんかん])から軽度(XLID±眼振および付加的な臨床的特徴)まで幅広いスペクトラムを有する[Moog et al 2015]。
これまでに、130人の患者(男性45人および女性85人)でCASK遺伝子病原性変異を同定されている[Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Takanashi et al 2012, Moog et al 2015, Dunn et al 2017, Hayashi et al 2017, Muthusamy et al 2017, Cristofoli et al 2018, Rama Devi et al 2019]。病態に関連する表現型の以下の記述は、これらの報告に基づいている。
女性
これまでに計85人のMICPCHの女性が報告されており、そのうちの最年長は25歳である。以下の自然史に関する情報は、Moog et al [2011]、Burglen et al [2012]、Hayashi et al [2012]、Takanashi et al [2012]の最近のレビューに基づいている。
MICPCH
頭囲
出生時の後頭前頭の周囲径(OFC)は、罹患女性の約3分の2で正常または正常下限の範囲にあり、その他は小頭症(OFC < -2 SD)である。小頭症は、例外なく最初の1年間に進行(OFC -3.5〜-10 SD)し、通常は出生4か月の間に進行する。
発達遅滞/知的障害(DD/ID)
罹患女性は、生後2~24か月の間に頚定と追視を獲得する。ほとんどの罹患女性は、生後7~36か月の間に独座ができるようになるが、独歩を獲得できるのは20~25%にすぎない(生後18~72か月の間)。
ほとんどは有意語がない。2語文を話すことができる者が1人いる。知的発達は、ほぼすべての女性で重度の障害があり、中等度の知的障害を呈する者もいる。
行動の表現型には、睡眠障害、手の常同運動、自傷行為がある。
神経学的特徴
(体軸性の)筋緊張低下、四肢の過緊張(痙性に進行する可能性がある)、およびジストニアまたはその他の運動障害が含まれる。様々なタイプのけいれん発作が約40%に認められ、発症は出生時から10歳までの間である。
MRIで観察される橋小脳低形成の重症度は、予後としての有用性はない[Moog et al 2011]。
MRI所見
- 小脳の軽度から重度のびまん性低形成を伴う橋小脳低形成は、大脳半球および小脳虫部に比例的に影響を及ぼす[Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Takanashi et al 2012] (図1)。脳橋と小脳は、進行性小頭症、知的障害、およびCASK遺伝子病原性変異を有する2人の女性で、正常形態を呈していることが報告されている[Cristofolietal2018]。
- 小脳半球は非対称的に影響を受けることがある。
- 脳橋低形成は、脳橋の隆起が相対的に軽度から重度の場合がある。
- 大脳と脳梁の比率が低い、正常または正常下限サイズの脳梁[Takanashietal 2010]
- 関連するMRI所見:大脳皮質前頭部脳回の数と複雑性の軽度減少と側脳室の軽度拡大 [Moog et al 2011]
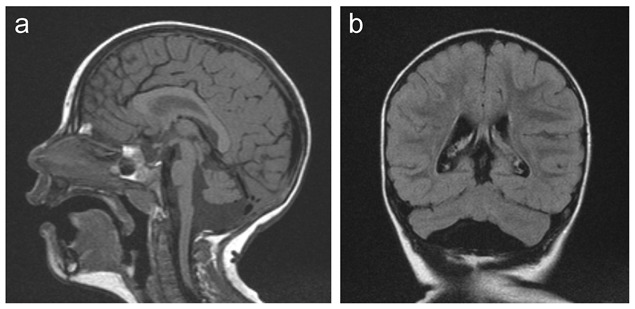
図1. MICPCHとCASK遺伝子のヘテロ接合性病原性変異を有する2歳6か月女児の脳MRI
- 矢状断は、隆起した脳橋を伴う軽度の橋小脳低形成を示している。脳梁は正常である。
- 冠状断は、大脳半球と小脳虫部に比例的に影響を及ぼす軽度の小脳低形成を示している("蝶"のような外観)。
他の所見
- 出生時の身長は正常。一般的に、4歳までには低身長である[Moog et al 2011, Takanashi et al 2012]。
- 脊柱側弯症が多くみられる。
- 様々な眼科所見があり、特に視神経低形成、網膜異常症、眼振、および斜視がありうる[LaConte et al 2019]。
- 罹患女性の約28%が感音性難聴である[Moog et al 2011, Burglen et al 2012, Takanashi et al 2012]。
- 先天性の臓器構造異常(例:腎/泌尿器系または心臓構造異常)はまれである;特定の異常が反復的に発生することはない。
- 最近のレビューでは、アーチ状の眉毛、広い鼻梁と鼻尖、小さいか短い鼻、人中が長いか上顎が突出している、小顎、耳が大きいといった特徴の顔面の表現型が報告されている。
罹患女性の死亡率は報告されていない。
X連鎖性知的障害(XLID)±眼振
ヘテロ接合体の大多数(典型的には、より重症な男性罹患者の血縁者で同定される)における臨床所見:
- 正常な知能、一部の女性のみで軽度から重度の知的障害
- 先天性眼振や斜視を含む正常から軽度の眼所見
- 軽度の振戦や欠神発作以外の神経学的症状はない
- MRI所見:正常または主に不明
男性
CASK遺伝子病原性変異を有する出生から59歳までの計45名の男性が報告されている[Moog et al 2015, Dunn et al 2017, Hayashi et al 2017, Muthusamy et al 2017, Rama Devi et al 2019]。
男性の表現型は、重症から軽症までの臨床的連続性があり、3つのグループに分類することができる[Moog et al 2015]。
重症てんかん性脳症を伴うMICPCH
頭囲
出生時、半数の患者でOFCは正常(下限)であった。残りの半数は、原発性小頭症(OFC <-2 SD)であった。軽度から重度の出生後の小頭症は、最初の数か月間に急速に進行した(OFC -2.7~-9 SD)。
DD/ID
すべての罹患男性は、重度から最重度のDD、またはそもそも発達がみられない。
神経学的特徴
早期・難治性のてんかん発作(大田原症候群 [Saitsu et al 2012]、ウエスト症候群 [Takanashi et al 2012]、ミオクロニーてんかん [Nakamura et al 2014])、バーストサプレッション・スパズム[Moog et al 2015]、運動過多 [Rama Devi et al 2019]などが挙げられる。
MRI所見
- 典型的な重度のびまん性橋小脳低形成
- 単純な脳回、皮質萎縮、髄鞘低形成
その他の所見
- 複数の(軽度の)先天異常が報告されている[Burglen et al 2012, Saitsu et al 2012, Moog et al 2015]。
- 心房中隔欠損、ファロー四徴症、水腎症 [Nakamura et al 2014, Moog et al 2015]。
死亡率
この表現型を有する男性は、周産期または早期に致死的となる可能性がある。罹患男性のうち1人は生後2か月で死亡しており[Rama Devi et al 2019]、1人は生後7か月で死亡しており、もう1人は生後21か月で死亡している[Moog et al 2015]。
重度の発達遅滞を伴うMICPCH
重度のてんかんを伴わないが、重度の発達遅滞とMICPCHの組み合わせは、6人の男性で報告されている。このグループの男性患者の表現型は、女性のMICPCHに匹敵するものである[Moog et al 2015, Hayashi et al 2017]。
- 頭囲 出生後の小頭症
- DD/ID 重症
- 神経学的特徴 男性1人に軽度の運動失調、別の男性にジストニア/ジスキネジアが報告された。てんかん発作の報告はない。
- MRI所見 程度の異なるびまん性橋小脳低形成
- その他の所見 眼振
- 死亡率 罹患男性1人が生後2週間で死亡した。
X連鎖性知的障害(XLID)±眼振
眼振および/または他の先天異常を伴う、または伴わない軽度から重度のXLIDが、合計29人の男性で報告されている[Moog et al 2015, Dunn et al 2017, Hayashi et al 2017]。
- DD/軽度から重度のID
- てんかん発作
- 先天性眼振、および斜視を含むその他の眼所見、軽度の視神経乳頭蒼白
脳MRIは少数の患者でのみ報告されており、橋小脳低形成は認められていない。
その他の所見は、小頭症、筋緊張低下、自閉症スペクトラム、行動の問題、振戦や不安定な歩行、感音性難聴、摂食困難、便秘、低身長、停留精巣、消化器や胃食道の合併症などがある。
遺伝子型-表現型の相関
女性では、MICPCHはCASK遺伝子のヘテロ接合性病原性機能喪失変異と関連している[Moog et al 2011、Burglen et al 2012、Hayashi et al 2012、Takanashi et al 2012、Hayashi et al 2017]。女性では、眼振の表現型を伴う、または伴わないXLIDは典型的にはCASK遺伝子病原性hypomorphic変異と関連している。
男性では、臨床的に区別可能な3つのグループは、CASK遺伝子病原性変異の分類に関連している[Moog et al 2015]:
- 最重症な表現型である、重症てんかん性脳症を有するMICPCHの男性におけるCASK遺伝子病原性変異の大部分は、生殖細胞系列で機能喪失変異である。
- MICPCHのグループでは、男性はCASK遺伝子機能喪失変異の体細胞モザイクか、またはヘミ接合性で部分的に浸透率のある変異である。
- 眼振を伴う、または伴わないXLID男性の最大のグループは、典型的には、ミスセンス変異およびスプライス変異を含むCASK遺伝子hypomorphic病原性変異を有する[Moog et al 2015]。
浸透率
MICPCH表現型(CASK遺伝子のヘテロ接合性病原性機能喪失変異に関連する)の浸透率は、これまでに報告された女性患者では完全浸透である。
CASK 遺伝子病原性変異の浸透率は、男性では完全浸透である。CASK遺伝子病原性変異のモザイクを持つ男性では、体細胞モザイクのレベルが臨床的多様性を決定する1つの因子である可能性がある。CASK遺伝子病原性hypomorphic変異のヘテロ接合体を持つ女性では、浸透率は不完全であり、臨床的多様性を有している。
命名法
FG症候群(FGS)様の表現型は、2つの家族の罹患男性における所見に基づいて、CASK異常症関連の表現型として明確に示唆されている[Piluso et al 2009, Dunn et al 2017]。しかしながら、MED12遺伝子の再発性病原性変異で発症するFGS1を除いて(MED12関連疾患を参照)、FGSは明確に定義されておらず、FGS4は表現型として認識できない。したがって、これらの家族に記載されている表現型は、眼振の有無にかかわらずXLIDに分類することがより適切である。
有病率
CASK異常症の有病率は不明である。少なくとも130人(男性45人、女性85人)のCASK
遺伝子病原性変異が報告されている。
遺伝的に関連する(アレル)疾患
GeneReviewに記載されている以外の表現型は、CASK遺伝子病原性変異と関連していることは知られていない。
鑑別診断
知的障害と脳橋および小脳低形成を伴う小頭症(MICPCH)
表2. MICPCHの鑑別診断で考慮すべき遺伝子
| 遺伝子 | 疾患 | MOI | 臨床的特徴 | 脳MRI所見 |
|---|---|---|---|---|
| SEPSECS TSEN15 TSEN2 TSEN34 VPS53 TSEN54 |
PCH2 | AR |
|
PCH2/PCH4の患者:
|
| TSEN54 | PCH4 | AR | 羊水過多、関節拘縮、重度の全身性クローヌス、中枢性呼吸不全は通常→新生児期死亡 | |
| ARX STXBP1 (>80遺伝子) 1 |
大田原症候群 | XL AD |
サプレッションバーストを伴う早期乳児てんかん性脳症 | 脳MRIで異常があるかどうかは不明 |
AD=常染色体優性; AR=常染色体劣性; MOI=遺伝形式; PCH=橋小脳低形成; XL=X染色体連鎖
- OMIMにおけるこの表現型に関連する遺伝子については、表現型シリーズ、早期乳児てんかん性脳症を参照
- CASK異常症では、大脳半球と小脳虫部のびまん性低形成に起因する「蝶」のパターンがみられる
X連鎖性知的障害(XLID)±眼振
眼振を伴うXLIDは、SLC16A2遺伝子のヘミ接合性病原性変異で発症するX連鎖性疾患のAllan-Herndon-Dudley症候群で認められることがある。これらの患者は、重度の知的障害、小頭症、神経学的特徴(痙縮、ジストニア、運動失調)、脊柱側弯症、大きな耳、その他の奇形を呈する。眼振は一部の患者で報告されている。
眼振を伴わないXLIDは、多くの遺伝子が非症候性および症候性XLIDの原因となることが知られているため、幅広い鑑別診断がある(OMIM表現型シリーズ: Nonsyndromic XLID, Syndromic XLIDを参照)。
臨床的マネジメント
初期診断後の評価
CASK異常症と診断された患者の疾患の程度とニーズを確立するためには、表3にまとめられた評価(診断につながった評価の一部として実施されていない場合)を行うことが推奨される。
表3. CASK異常症患者で初期診断に続いて推奨される評価
| 器官系 | 評価 | コメント |
|---|---|---|
| 神経 | 神経学的評価 | まだ行われていない場合は、脳MRIと脳波を含む |
| 発達 | 発達評価 |
|
| 精神/ 行動 |
神経精神学的評価 | 12歳以上の患者:睡眠障害、ADHD、不安、および/ またはASDを示唆する形質を含む行動上の懸念へのスクリーニング |
| 筋骨格系 | 整形外科/理学療法・リハビリテーション/PT/OT 評価 |
|
| 消化器/摂食 | 消化器科/栄養科/ 摂食チーム評価 |
|
| 眼 | 眼科的評価 | 眼振、視神経低形成、網膜症、および斜視の評価 |
| 聴力 | 聴力検査 | 難聴の評価 |
| 心血管 | 心臓超音波検査 | 稀だが心臓構造異常を評価する |
| 泌尿器 | 腎臓超音波検査 | 稀だが腎臓/泌尿器構造異常を評価する |
| その他 | 臨床遺伝学者・遺伝カウンセラーとの相談 | 遺伝カウンセリングを含む > |
| 家族支援/リソース |
|
ADHD=注意欠陥・多動性障害; ASD=自閉症スペクトラム障害; OT=作業療法士; PT=理学療法士
症状に対する治療
表4. CASK異常症患者の症状に対する治療
| 症状 | 治療 | 考慮すべきこと/その他 |
|---|---|---|
| DD/ID | 発達遅滞/知的障害の管理を参照 | |
| てんかん | 経験豊富な神経内科医によるAEDを用いた標準治療 |
|
| 体重増加不良 | 摂食療法;持続的な摂食問題には 胃瘻チューブの挿入が必要な場合がある |
嚥下障害の臨床徴候や症状がある場合は、臨床的摂食評価および/または嚥下造影検査の閾値は低い |
| 痙縮 | 整形外科/理学療法・リハビリテ ーション/PT/OT ストレッチで関節拘縮や転倒を防ぐ |
ポジショニング・移動機器、障害者用 駐車プラカードの必要性を考える |
| 視野異常・斜視 | 眼科医が推奨する標準治療 | 早期介入または学区によるコミュニティビジョンサービス |
| 聴覚 | 補聴器が役に立つかもしれない | 早期介入または学区によるコミュニティ聴覚サービス |
| 家族・コミュニティ |
|
|
AED=抗てんかん薬; DD=発達遅滞; ID=知的障害; OT=作業療法; PT=理学療法
- 一般的なてんかん発作に関する保護者/介護者への教育が適切である。てんかんと診断された子どもへの非医学的介入および対処戦略に関する情報は、てんかんとわが子のツールキットを参照。
発達遅滞/知的障害の管理問題
以下の情報は、米国の発達遅滞/知的障害のある患者に対する一般的な管理上の推奨事項を示している。標準的な推奨事項は、国によって異なる場合がある。
0-3歳.
作業療法、理学療法、言語療法、食事療法だけでなく、乳幼児のメンタルヘルスサービス、特別教育者、感覚障害の専門家といった早期介入プログラムへの参加が推奨されている。米国では、早期介入はすべての州で利用可能な連邦基金によるプログラムであり、個々の治療ニーズを対象とした在宅サービスを提供している。
3-5歳.
米国では、地元の公的な学区を通して、発達上のプレスクールが推奨される。配備前に、必要なサービスと治療法を決定するために評価が行われ、運動、言語、社会性、認知の遅れが認められる場合には、個別教育プログラム(IEP)が展開される。 早期介入プログラムは通常、この移行を支援する。発達上のプレスクールはセンター制だが、医学的に不安定で通うことができない子どもたちには、家庭でのサービスが提供される。
全年齢.
発達小児科医との相談は、適切なコミュニティ、州と教育機関の関与を確保し、生活の質を最大にする上で両親を支援するために推奨される。
- IEPサービス:
- IEPは、資格のある子どもたちに対して、特別に設計されたインストラクションと関連するサービスを提供する。
- IEPサービスは、内容変更の必要性を判断するために、毎年見直しが行われる。
- 特別教育法で定められているように、子どもたちは学校で可能な限り制限の少ない環境に置かれ、可能な限りかつ適切な場合には、一般教育に参加するできである。
- 視覚と聴覚の専門家は、こどものIEPチームの一員として、こどもが学術的な教材を使用できるようにサポートしなければならない。
- 理学療法士、作業療法士、言語聴覚士による支援は、こどもが学術的な教材を利用するために必要な範囲内で、IEPの中で提供される。それ以上の場合は、罹患者のニーズに基づいた個人的な支持療法を検討することができる。支持療法の種類については、発達小児科医が具体的な推奨を行うことができる。
- 子どもが10代になると、成人期への移行計画について話し合い、IEPに盛り込む必要がある。IEPサービスを受けている場合、公立学校区は21歳までサービスを提供することが義務付けられている。
- 504計画(セクション504:障害に基づく差別を禁止する米国連邦法)では、教育現場の適応や改良を必要とする者のために、教室の前の方に座席を置く、補助技術機器の使用、スクライビング、授業と授業の間の時間の延長、課題の修正、教科書の文字を拡大することなどを検討することができる。
- 発達障害管理局 (DDA) への登録が勧められる。DDAは米国の公的機関であり、資格のある者にサービスとサポートを提供している。資格は州によって異なるが、一般的には診断名および/または関連する認知/適応障害の程度により決定される。
運動機能障害
粗大運動障害
- 可動性を最大限に高め、後に発症する整形外科的合併症 (関節拘縮、脊柱側弯症、股関節脱臼など) のリスクを軽減するために、理学療法が推奨される。
- 必要に応じて耐久性のある医療機器およびポジショニング機器の使用を考慮する (例:車椅子、歩行器、入浴椅子、装具、適応ベビーカー)。
- 筋緊張低下またはジストニアを含む筋緊張異常については、バクロフェン、チザニジン、ボトックス®、抗パーキンソン薬、または整形外科的処置による管理といった手助けを得るため、適切な専門家への支援依頼を検討する。
微細運動障害
食事、身だしなみ、着衣、および筆記などの日常生活機能に影響を及ぼす微細運動の障害に対しては、作業療法が推奨される。
口腔機能障害
毎回の診察において口腔の運動機能障害を評価し、食事中の窒息/嘔気、体重増加不良、頻回の呼吸器疾患罹患、または他に説明のつかない摂食拒否について、臨床的な摂食評価および/または嚥下造影検査を実施する必要がある。口から食べても安全であると仮定して、協調性や感覚に関連した摂食の問題を改善するために、摂食療法 (通常は作業療法士または言語聴覚士による) を行うことが推奨される。安全のために、食事にとろみをつけたり、冷やしたりすることができる。摂食機能の障害が重度の場合には、経鼻胃管挿入または胃瘻造設術が必要になることがある。
コミュニケーションの問題
表出性言語障害を有する患者に対しては、代替コミュニケーション手段(例えば、拡大代替コミュニケーション[AAC])の評価を検討すべきである。AACの評価は、この分野の専門知識を持つ言語聴覚士が行うことができる。AAC評価では、認知能力や感覚障害を考慮し、最も適切なコミュニケーションの形態を決定する。AAC装置には、画像交換コミュニケーションのようなローテクなものから、音声生成装置のようなハイテクなものまである。一般的に信じられていることとは逆に、AAC装置は言語発達を妨げるものではなく、多くの場合、言語発達を改善することができる。
社会的/行動的懸念
子どもは、応用行動分析(ABA)を含む自閉症スペクトラム障害の治療に使用される介入の資格があり、その恩恵を受けることができる。ABA療法は、個々の子どもの行動的、社会的、および適応的な長所と短所を対象としており、通常認定された行動分析者と一対一で実施される。
発達小児科医との相談は、適切な行動管理計画を通して両親を指導し、注意欠陥・多動性障害(ADHD)に対しては必要に応じて処方を提供するのに役立つ場合がある。
重度の他害行動や自傷行動についての問題は、小児精神科医が対応することもある。
サーベイランス
表5. CASK異常症患者に対して推奨されるサーベイランス
| 対象/関連臓器 | 評価項目 | 頻度 |
|---|---|---|
| 摂食 |
|
受診毎 |
| 神経 | てんかん発作が臨床的に疑われるかの監視 | 受診毎 |
| てんかん発作、調子の変化、運動障害などの新規症状の評価 | ||
| 発達 | 発達の進捗状況と教育の必要性の監視 | 受診毎 |
| 精神/行動 | 不安、注意、攻撃的または自傷行為に対する行動評価 | |
| 筋骨格 | 理学療法医、OT/PTによる運動能や自力で可能な技能の評価 | 受診毎 |
| 眼 | 眼科的評価 | 1年に1回 |
| 聴覚 | 聴力検査 | 1年に1回 |
| その他 | 家族への社会的支援 (例:緩和ケア/レスパイトケア、 訪問看護、その他の地域による支援) とケア調整の必要性の評価 |
受診毎 |
OT=作業療法士; PT=理学療法士
リスクのある血縁者の評価
遺伝カウンセリングの目的でリスクのある血縁者を検査することに関する問題については、遺伝カウンセリングを参照。
研究中の治療法
広い範囲の疾患と健康状態に対する臨床研究情報にアクセスするためには、米国のClinicalTrials.govおよび欧州のEU Clinical Trials Registerを検索のこと。注釈:この疾患における臨床試験はないかもしれない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
眼振を伴う、または伴わない、知的障害やMICPCH、XLIDは、CASK遺伝子病原性変異によって発症し、X染色体連鎖の遺伝形式である。
CASK異常症の発端者の家族へのリスクは、表現型(すなわち、MICPCHまたはXLID±眼振)に依存する。
家族構成員のリスク
- MICPCH
MICPCH女性の多くは、孤発例(すなわち、1家系に1人だけ発症)であり、 CASK遺伝子の新生病原性変異の結果として発症する。しかし、(可能性は低いが)MICPCH女性が、体細胞および/または生殖細胞系列モザイクを持つ母親または父親からCASK遺伝子病原性変異を受け継いだ可能性もある。
もし親が無症状であれば、MICPCH表現型は完全浸透であるようなので、親が病原性変異を有する可能性は低いと考えられる。しかし、(可能性は低いが)発端者の親が、体細胞および/または生殖細胞系列モザイクを有している可能性はある。
- XLID±眼振
XLID±眼振の女性は、新生病原性変異または母親から受け継いだ病原性変異の結果として発症することがある(ヘミ接合性の男性は罹患するので、XLID±眼振の女性が父親から病原性変異を受け継いだ可能性は低い)。
- 母親(場合によっては父親)の分子遺伝学的検査により、CASK遺伝子病原性変異が遺伝したかどうか判断するのに役立つかもしれない。
- 注:CASK遺伝子病原性変異がどちらの親からも検出されない場合、その病原性変異は多くの場合、発端者に新規に発生した可能性が高い。もう一つ考えられる説明としては、生殖細胞(または体細胞と生殖細胞)にモザイクを持つ親から病原性変異を受け継いだということも考えられる。親の白血球のDNA検査では、体細胞モザイクのすべての例を検出できない場合がある。
男性発端者の両親
- CASK異常症の男性の父親は、疾患を有していない、あるいはCASK遺伝子のヘミ接合性病原性変異である;したがって、さらなる評価/検査を必要としない。
- MICPCH
重度のてんかん性脳症を伴うか否かにかかわらず、MICPCH男性の多くは、孤発例(すなわち、1家系に1人だけ発症)であり、新生突然変異または体細胞モザイクのCASK遺伝子病原性変異の結果として発症する。
もし、発端者の母親が無症状であれば、MICPCH表現型は完全浸透であるようなので、ヘテロ接合性病原性変異である可能性は低いと考えられる。しかし、(可能性は低いが)母親が体細胞モザイクおよび/または生殖細胞系列モザイクを有する可能性はある。(CASK遺伝子欠失のモザイクは、無症候性の罹患男性の他の一人で報告されている[Saitsu et al 2012])
- XLID±眼振
罹患者が男性のみの家族の場合、母親がヘテロ接合体*であるか、または罹患男性が新生病原性変異を有している可能性がある(XLIDの家族を評価した結果、ほとんどの男性が同定されているため、どのくらいの数の罹患男性が孤発例かは不明である)。複数の罹患者がいる家族では、罹患男性の母親は絶対的ヘテロ接合体である。
母親の遺伝学的状態を確認するために、母親の分子遺伝学的検査を行うことが推奨される。(注:女性に2人以上の罹患している子供がいて、他に罹患している親族がおらず、白血球のDNAからCASK遺伝子病原性変異が検出できない場合、その女性は生殖細胞系列モザイクである可能性が高い)。
* ヘテロ接合体の女性は典型的には無症状であるが、軽度から重度の知的障害であることがあり、眼球機能の欠如、欠神発作、および/または振戦を合併することもしないこともある
- 注:CASK遺伝子病原性変異が母親から検出されない場合、その病原性変異はほとんどの場合、発端者に新規に発生した可能性が高い。別の可能性としては、生殖細胞系列モザイク(または体細胞モザイクと生殖細胞系列モザイク)を有する母親から病原性変異を受け継いだということも考えられる。母親の白血球DNAの検査では、体細胞モザイクのすべての例を検出できない場合がある。
女性発端者の同胞 同胞へのリスクは、両親の遺伝学的状態に依存する。
- MICPCH
発端者が孤発例の場合、母親(男性と女性の同胞にリスクがある)または父親(女性の同胞にリスクがある)に生殖細胞系列モザイクの可能性があるため、同胞への再発リスクは低いようにみえるが、一般集団よりはリスクが高い。
- XLID±眼振
発端者の母親がCASK 遺伝子病原性変異を有する場合、各妊娠で受け渡す確率は50%である(この病原性変異を受け継いだ男性は罹患する。病原性変異を受け継いだ女性は、典型的には無症状であるが、軽度から重度の知的障害であることがあり、眼球機能の欠如、欠神発作、および/または振戦を合併することもしないこともある)。CASK遺伝子病原性変異が母体の白血球DNAから検出できない場合、親の生殖細胞系列モザイクの可能性があるため、同胞のリスクは一般集団よりは高い。
男性発端者の同胞 発端者の同胞へのリスクは、母親の遺伝学的状態に依存する。
- MICPCH
発端者が孤発例の場合、母親に生殖細胞系列モザイクの可能性があるため、同胞への再発リスクは低いようにみえるが、一般集団よりはリスクが高い。
- XLID±眼振
母親がCASK遺伝子のヘテロ接合性病原性変異を有する場合、各妊娠で受け渡す確率は50%である(この病原性変異を受け継いだ男性は罹患する。病原性変異を受け継いだ女性は、典型的には無症状であるが、軽度から重度の知的障害であることがあり、眼球機能の欠如、欠神発作、および/または振戦を合併することもしないこともある)。CASK遺伝子病原性変異が母親の白血球DNAから検出できない場合、母親の生殖細胞系列モザイクの可能性があるため、同胞のリスクは一般集団よりは高い。
女性発端者の子
- MICPCH 現在までのところ、MICPCH女性の生殖についての報告はない。
- XLID±眼振 XLID±眼振の女性は、50%の確率でCASK遺伝子病原性変異を子供に受け渡す。
男性発端者の子
現在までのところ、CASK異常症男性の生殖についての報告はない。
他の家族
男性発端者の母方の叔母および母方の従兄弟は、病原性変異を有するリスクがあるかもしれない。注:分子遺伝学的検査は、新規病原性変異が生じた家族構成員を特定することができるかもしれない。この情報は、親戚の遺伝学的リスク状態を判断する助けになりうる。
遺伝カウンセリングに関連した問題
家族計画
- 遺伝的リスクの決定や出生前検査の適応に関する議論の最適な時期は妊娠前である。
- ヘテロ接合性変異を有する(有症状あるいは無症状)、あるいは有するリスクがある若年成人に対して遺伝カウンセリング(子孫への潜在的なリスクや生殖の選択肢の議論を含む)を提供することは適切である。
DNAバンキングは、(通常は白血球から抽出された)DNAを将来の使用のために保存しておくものである。検査の手法や遺伝子、アレル変異および疾患に対する我々の理解が将来進歩するかも知れないので、罹患者のDNAの保存は考慮すべきである。
出生前検査および着床前診断
罹患した家族の中にCASK遺伝子病原性変異が同定されると、CASK異常症のリスクのある妊娠のための出生前検査と着床前診断を検討することがある。
医療の専門家の間や家族内においても、出生前診断に対する考え方の相違が存在しうる。ほとんどの施設では出生前診断をするかどうかは個人の問題ととらえているが、 これらの問題を議論することは有益である。
資源
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- CASK Kinder-und Lebenshilfe e. V.
ドイツ
Phone: +49 (0) 6154 8018537
Email: info@cask-kinder-lebenshilfe.de
www.cask-kinder-lebenshilfe.de - American Association on Intellectual and Developmental Disabilities (AAIDD)
501 3rd Street Northwest
Suite 200
Washington DC 20001
Phone: 202-387-1968
Fax: 202-387-2193
Email: sis@aaidd.org
www.aaidd.org - Medline Plus
Intellectual Disability
• National Center on Birth Defects and Developmental Disabilities
Phone: 800-232-4636 (フリーダイヤル)
Email: cdcinfo@cdc.gov
Facts About Intellectual Disability
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A. CASK異常症の遺伝子とデータベース
| 遺伝子 | 染色体座位 | タンパク質 | Locus-Specific Databases | HGMD | ClinVar |
|---|---|---|---|---|---|
| CASK | Xp11.4 | Peripheral plasma membrane protein CASK | CASK @ LOVD | CASK | CASK |
データは、以下の標準的な参考文献を編集したものである:HGNCの遺伝子;OMIMの染色体座位、UniProtによるタンパク質。リンク先のデータベース (Locus Specific, HGMD, ClinVar) の説明は、 ここをクリック。
表B. OMIMに登録されているCASK異常症(OMIMで全てを参照のこと)
| 300172 | CALCIUM/CALMODULIN-DEPENDENT SERINE PROTEIN KINASE; CASK |
| 300422 | FG SYNDROME 4; FGS4 |
| 300749 | MENTAL RETARDATION AND MICROCEPHALY WITH PONTINE AND CEREBELLAR HYPOPLASIA; MICPCH |
分子学的病因
ASK遺伝子は、膜結合型グアニル酸キナーゼ(MAGUK)ファミリーのマルチドメインタンパク質であるカルシウム/カルモジュリン依存性セリンプロテインキナーゼ(CASK)をコードしている。CASKは様々な組織で発現しているが、脳の様々な領域に広く分布している。
CASKは下記を含む。
- N末端カルモジュリン依存性プロテインキナーゼ(CamK)ドメイン
- 2つのL27(L27.1とL27.2)ドメイン
- PSD-95/discs large/ZO-1(PDZ)ドメイン
- srcホモロジー3 (SH3)
- C末端のグアニル酸キナーゼ(GK)ドメイン
CASKは脳の発達と機能において重要な役割を果たしている。CASKは、(1)シナプス前の組織化と神経伝達物質放出の調節、(2)樹状突起の形態維持とシナプス後へのグルタミン酸受容体の輸送、(3)皮質発達に関与する遺伝子の転写調節によって、シナプスの形成と活動を制御している[Hsueh 2006, Hsueh 2009]。
疾患発症メカニズム
MICPCH女性および、重度のてんかん性脳症を伴う、または伴わないMICPCH男性におけるCASK遺伝子病原性変異の大部分は、ナルアレルと予測され、重度の表現型と関連している[Najm et al 2008, Moog et al 2011, Burglen et al 2012, Hayashi et al 2012, Moog et al. 2015, Hayashi et al 2017]。ヘミ接合性の病原性機能喪失変異を有する男性は、女性よりも重症である。
MICPCH男性で同定されたいくつかのヘテロ接合性ミスセンス変異は、相互作用パートナーであるMint1またはneurexinへのCASKの結合を特異的に損なう。この障害が、女性では重度の表現型を発症するのに十分な根拠となりうるかどうかは疑問が残されたままである[LaConte et al 2018, LaConte et al 2019]。
眼振を伴う、または伴わないX連鎖性知的障害を有する男性(およびまれに女性)におけるCASK遺伝子のhypomorphic病原性変異は、主にミスセンス変異およびスプライス変異である。これらの変異は、CASKの他の機能をそのまま残しつつ、CASKタンパク質の特定の機能を阻害する可能性がある[Moog et al 2015]。
更新履歴:
-
GeneReviews著者:Ute Moog, MD and Kerstin Kutsche, PhD
日本語訳者:西村直人(防衛医科大学校病院 小児科)、黒澤健司(神奈川県立こども医療センター 遺伝科)
GeneReviews最終更新日: 2020.5.21. 日本語訳最終更新日: 2021.5.21.[ in present]
![]()