脳クレアチン欠乏症候群
(Cerebral Creatine Deficiency Disorders)
[Synonyms: Creatine Deficiency Disorders]
GeneReviews著者: Saadet Mercimek-Andrews, MD, PhD, FCCMG, FRCPC,Gajja S Salomons, PhD
日本語訳者:和田 敬仁 (京都大学大学院医学研究科ゲノム医療学講座)
GeneReviews最終更新日: 2022.2.10. 日本語訳最終更新日: 2022.3.14.
原文 Creatine Deficiency Disorders
要約
疾患の特徴
クレアチン代謝および輸送の先天性異常であるクレアチン欠乏症 (CDD) は、クレアチン生合成障害のグアニジノ酢酸メチルトランスフェラーゼ (GAMT) 欠損症、L-アルギニン:グリシンアミジノトランスフェラーゼ (AGAT) 欠損症、および、クレアチントランスポーター (CRTR) 欠損症の3つの疾患から構成されている。
- GAMT欠損症の臨床症状は、生後3ヶ月から2年の間に発症し(約130名が報告)、発達の遅れに加え、大多数がてんかんを持ち、行動障害(例:多動、自閉症または自傷行為)を発症し、約30%が運動障害である。
- AGAT欠損症は16名で報告されているが、てんかんや運動障害を有する者はいない。
- 男性におけるCRTR欠損症の臨床所見は(約130人報告されており)、発達遅延に加え、てんかん(発作型が多様で難治性の場合もある)、行動障害(注意欠陥や多動性、自閉症の特徴、衝動性、社会不安など)、筋緊張低下、(頻度は低いが)運動障害などがある。便秘を伴う不十分な体重増加や、心電図 (EKG) 上の QTc 延長も報告されている。4歳までは軽度から中等度の知的障害が一般的であるが、CRTR欠損症の成人男性の大多数は、重度の知的障害を有すると報告されている。CRTR欠損症のヘテロ接合体の女性は、典型的には、無症候性または軽度の知的障害であるが、男性の表現型に似たより重度の表現型が報告されている。
診断・検査
示唆的な臨床所見を呈し、分子遺伝学的検査により同定された、GAMTまたはGATMの両方のアレルの病原性バリアント、あるいは、SLC6A8のヘミ接合型(男性)またはヘテロ接合型(女性)の病原性バリアントをもつ発端者において、CDDの診断は確立される。
臨床的マネジメント
症状に対する治療:
GAMT欠損症およびAGAT欠損症は、脳内クレアチン濃度を補充するために、クレアチン モノハイドレートを経口投与することで治療を行う。GAMT欠損症の治療には、オルニチンの補充とアルギニンまたはタンパク質の食事制限が必要である。CRTR欠損症は、クレアチン モノハイドレートの経口投与とアルギニンおよびグリシンの補給で治療を行う。発達の遅れ、知的障害、および行動の問題は、個別の教育および治療プログラムによって管理され、てんかんおよび運動障害は、適切な専門家により標準的な方法で治療を行う。
サーベイランス:
クレアチンモノハイドレートを投与された患者では、クレアチン関連腎症の可能性を検出するために、インビボ1H-MRS による脳内クレアチンレベルの定期的な測定と、腎機能の年次測定が、根拠とされている。発育および神経学的な評価は、各診療所で行うことが推奨される。
リスクのある親族の評価:
生化学的あるいは分子遺伝学的検査により、CDDのリスクのある新生児を早期に診断することで、早期診断と治療が可能になる。
遺伝カウンセリング
GAMT欠損症(GAMTの病原性バリアントに起因する)およびAGAT欠損症(GATMの病原性バリアントにに起因する)は、常染色体潜性(劣性)遺伝する。CRTR欠損症(SLC6A8の病原性バリアントに起因する)は、X連鎖的に遺伝する。
- 常染色体潜性(劣性)遺伝:両親ともにGAMTまたはGATMの病原性バリアントのヘテロ接合であることが知られている場合、罹患者の兄弟姉妹は、受胎時に25%の確率で罹患し、50%の確率で無症候性保因者となり、25%の確率で無症候性で保因者でないことが分かっている。発症した家族においてGAMTまたはGATMの病原性バリアントが同定されると、分子遺伝学的保因者検査、出生前および着床前遺伝子検査が可能となる。
- X連鎖遺伝:SLC6A8病原性バリアントをヘテロ接合で持つ母親は、各妊娠時に50%の確率で病原性バリアントを伝達する。病原性バリアントを受け継いだ息子は罹患し、病原性バリアントを受け継いだ娘はヘテロ接合体となり、障害に関する臨床所見を発現する可能性がある。発症した家族においてSLC6A8病原性バリアントが同定されると、女性のヘテロ接合体を特定するための分子遺伝学的検査、出生前および着床前遺伝子検査が可能となる。
GeneReviewスコープ
| クレアチン欠乏症:以下の表現型で構成される。 |
|---|
|
診断
クレアチン欠乏症は、クレアチン代謝および輸送の先天性異常であり、その構成を、以下に記す。
- 2つのクレアチン生合成の欠陥(いずれも常染色体潜性(劣性)遺伝):
- グアニジノ酢酸メチルトランスフェラーゼ (GAMT) 欠損症
- L-アルギニン:グリシンアミジノトランスフェラーゼ (AGAT) 欠損症
- クレアチントランスポーター欠陥1件(X連鎖遺伝):クレアチントランスポーター (CRTR) 欠損症
疾患を疑う所見
以下のような、臨床的所見、生化学的所見、画像所見、家族の病歴を有する者は、CDDを疑う必要がある。
臨床所見
- 発育遅延
- 認知機能障害または知的障害
- 筋緊張低下
- 発作または難治性てんかん
- 運動障害(舞踏病 - アテトーゼ、ジストニアなど)
- 行動上の問題(注意欠陥/多動性障害、自閉症スペクトラム障害、攻撃的な行動など)
生物学的所見 [van de Kampら 2014, Mørkridら 2015]
- GAMT欠損症:尿、血漿、または脳脊髄液 (CSF) のグアニジノ酢酸 (GAA) レベルが上昇し、尿、血漿、またはCSFのクレアチンおよびクレアチニンレベルが、低いまたは低正常の場合
- AGAT欠損症:尿、血漿、またはCSF中のGAAレベルが低く、尿、血漿、またはCSF中のクレアチンおよびクレアチニンレベルが、低いまたは低正常の場合
- CRTR欠損症:男性では、尿中のクレアチン / クレアチニン比が上昇する。女性では、尿中のクレアチン / クレアチニン比が、正常または軽度に上昇することがある。
画像所見
- プロトン磁気共鳴分光法 (1H-MRS) により、GAMT欠損症、AGAT欠損症の全患者とCRTR欠損症の男性で、脳内のクレアチンピークがない、または著しく減少していることが明らかになる[van de Kampら 2014]。
- 1H-MRSにより、X連鎖性CRTR欠損症のヘテロ接合体女性において、脳内のクレアチンピークの部分的喪失、または正常レベルが明らかになる [van de Kampら 2011]。
家族歴
- GAMT欠損症とAGAT欠損症:常染色体潜性(劣性)遺伝と一致する(例;同胞の罹患、あるいは、両親の近親婚など)。家族歴がなくても、診断を除外するものではない。
- CRTR欠損症:X連鎖遺伝と一致する(例;男性から男性への遺伝はない、母方の叔父または男性同胞が罹患、女性同胞または母親が正常から軽度の罹患など)。家族歴がなくても、診断を除外するものではない。
診断の確立
CDDの診断は、分子遺伝学的検査において、GAMTまたはGATMの両方のアレルの病原性バリアント、あるいはSLC6A8のヘミ接合体(男性)またはヘテロ接合体(女性)の病原性バリアントを同定することにより、臨床所見が示唆された発端者で確立される。
注記:(1) 意義不明の両方のアレルのGAMTバリアントまたはGATMバリアントの同定(または、既知の病原性バリアント1つと意義不明のバリアント1つの同定)は、GAMTまたはAGAT欠損症の診断を確定または除外するものではない。
これらの患者では、GAMTまたはAGAT欠損症の生化学的診断を確定するために、典型的な脳の1H-MRSおよび異常な尿や血漿、またはCSFのGAAおよびクレアチンレベルの異常、線維芽細胞またはリンパ球におけるGAMTおよびAGAT活性測定が必要とされるであろう。(2) ヘミ接合体(男性)またはヘテロ接合体(女性)の意義不明のSLC6A8バリアントの同定は、CTR欠損症の診断を確定または除外するものではない。これらの患者では、生化学的診断を確定するために、脳の1H-MRSの異常と男性の尿クレアチン/クレアチニン比の異常値が必要となる。男性におけるCRTR欠損症の生化学的診断を確定するためには、患者の培養皮膚線維芽細胞におけるクレアチン取り込みが必要となる。
診断検査アルゴリズムは、示唆的な臨床、生化学、および/または画像所見、および/または脳1H-MRSにおけるクレアチンレベルの低下が疑われる患者における分子遺伝学的検査の指針として有用である(図1参照)。
分子遺伝学的検査のアプローチには、表現型に応じて、標的遺伝子検査(単一遺伝子検査、てんかん、知的障害、または自閉症スペクトラム障害のための多遺伝子パネル)と包括的ゲノム検査(エクソームシーケンス、ゲノムシーケンス)の組み合わせが含まれることがある。
特定の遺伝子検査では、生化学的所見や画像所見から、どの遺伝子が関与しているかを、臨床医は判断する必要があるが、多遺伝子パネル、あるいは、包括的ゲノム検査では、その必要はない。示唆的所見に記載された特徴的な生化学的所見や画像所見を有する患者は、遺伝子検査(選択肢1参照)で診断される可能性が高く、一方、知的障害やてんかんを伴う他の多くの遺伝性疾患と区別がつかない表現型を有する患者は、遺伝学的検査(選択肢2参照)で診断される可能性が高くなる。
オプション 1
単一遺伝子検査:診断検査アルゴリズムから、予測される遺伝子の配列解析を最初に行い、遺伝子内の小さな欠失 / 挿入、ミスセンス、ナンセンス、スプライスサイトのバリアントを検出する。注記:使用するシーケンシング方法によっては、シングルエクソン、マルチエクソン、または全遺伝子欠失 / 重複が検出されない場合がある。使用したシーケンシング方法により、バリアントが1つだけ(GAMTまたはGATMの場合)、または全く(3遺伝子のいずれについても)検出されなかった場合、次のステップとして、エクソンおよび遺伝子全体の欠失または重複を検出するための、遺伝子ターゲット欠失 / 重複解析が行われる。
(翻訳者注;脳クレアチン欠乏症の3疾患に対する遺伝子検査はかずさ遺伝子検査室に依頼することが可能であり、令和4年度4月より保険収載される予定である。)
GAMT、GATM、SLC6A8、その他関心のある遺伝子(鑑別診断を参照)を含むてんかん、知的障害、自閉症スペクトラム障害の多遺伝子パネルが、基礎となる表現型を説明しない、遺伝子における意義不明のバリアントや、病原性バリアントの同定を制限しつつ、症状の遺伝的原因を特定する可能性が最も高いと考えられる。注記:(1) パネルに含まれる遺伝子と、各遺伝子に使用される検査の診断感度は、検査室によって異なり、時間の経過とともに変化する可能性がある。(2) 多遺伝子パネルの中には、このGeneReviewで論議されている疾患と、関連性のない遺伝子が含まれている場合がある。(3) いくつかの検査施設では、パネルのオプションとして、検査施設がデザインしたカスタムパネルや、臨床医が指定した遺伝子を含む表現型に、焦点を当てたカスタムエクソーム解析が含まれることがある。(4) パネルに使用される方法には、配列解析、欠失 / 重複解析、および/または他の非配列ベースの検査が含まれる場合がある。
マルチジーンパネルの紹介は、こちらを参照する。遺伝子検査を発注する臨床医のための、より詳細な情報は、こちらで確認できる。
オプション 2
包括的なゲノム検査では、臨床医がどの遺伝子が関与している可能性が高いかを判断する必要はない。エクソームシーケンスが最も一般的に使用されているが、ゲノムシーケンスも可能である。
包括的なゲノム検査の紹介は、こちらを参照する。ゲノム検査を発注する臨床医のためのより詳細な情報は、こちらで確認できる。
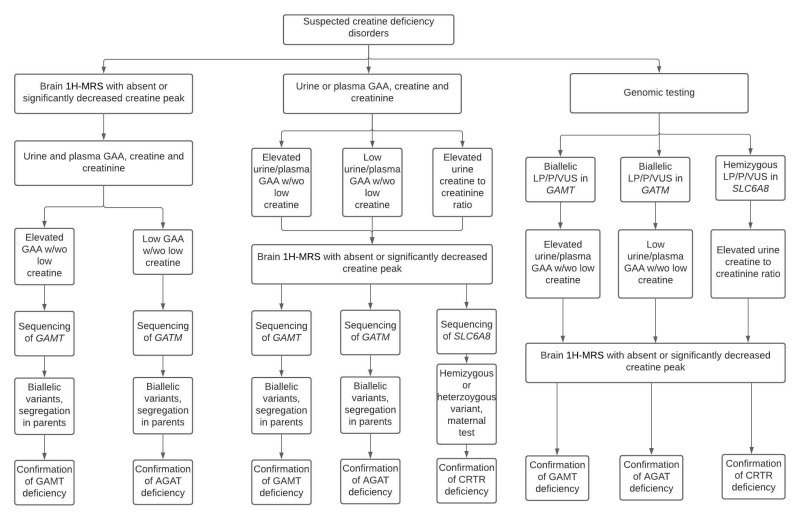
図 1. クレアチン欠乏症の診断アルゴリズム。注記:SLC6A8欠損症の女性では、尿中クレアチン/クレアチニン比および培養皮膚線維芽細胞におけるクレアチン取り込み試験は、しばしば有益ではない。そのため、この疾患の女性の診断には、分子遺伝学的検査が望ましい方法である[van de Kampら 2011]。
GAA = グアニジノ酢酸
表 1. クレアチン欠乏症で使用される分子遺伝子検査
| 遺伝子 1, 2 | 遺伝子の病原性バリアントに起因するCDDの割合 3 | 病原性バリアントの割合 3, 4方法によって検出可能 | |
|---|---|---|---|
| シーケンス分析 5 | 標的遺伝子の欠損/重複解析 6 | ||
| GAMT | 33% (20%) | ~100% | 不明 7 |
| GATM | 3% (8%) | ~100% (94%) | 不明 (6%) |
| SLC6A8 | 64% (72%) | ~95% (100%) | ~5% (0%) |
- 遺伝子は、アルファベット順に記載されている。
- 染色体座位とタンパク質については、表A.遺伝子とデータベースを参照する。
- パーセントは著者自身のデータベース / LOVD (SLC6A8, GATM, GAMT) のファミリー数に基づいており、() 内のパーセントは、異なる場合は Human Gene Mutation Database [Stensonら 2020] の購読制プロフェッショナルビューに基づいている。
- これらの遺伝子で検出されたバリアントについては、Molecular Geneticsを参照する。
- 配列解析では、良性、良性の可能性が高い、意義が不明、病原性の可能性が高い、または病原性バリアントが検出される。バリアントには、遺伝子内の小さな欠失や挿入、ミスセンス、ナンセンス、スプライスサイトのバリアントが含まれることがある。通常、エクソンや遺伝子全体の欠失や重複は検出されない。配列解析の結果を解釈する際に考慮すべき点については、こちらを参照する。
- 遺伝子欠損/重複解析は、遺伝子内の欠損や重複を検出する。定量PCR法、long-range PCR法、multiplex ligation-dependent probe amplification (MLPA)、シングルエクソン欠失・重複検出用の遺伝子ターゲットマイクロアレイなど、様々な手法が用いられる。
- 遺伝子ターゲットの欠失・重複解析の検出率に関するデータはない。
臨床的特徴
臨床的説明
発達遅延、認知機能障害、知的障害は、3つのクレアチン欠乏症 (CDD) に共通するものである。3つの欠損症の比較は、表2を参照する。詳細を以下の表に記す。
表 2. クレアチン欠乏症:選択した特徴による表現型の比較
| 特徴 | GAMT欠損症 1 | AGAT欠損症 2 | CRTR欠損症 3 |
|---|---|---|---|
| DDと認知機能障害4 | ●●● | ●●● | ●●● |
| 音声言語障害 | ●● | ●● | ●● |
| 発作 | ●● | ● | ●● |
| てんかん | ●● | NR | ●● |
| 行動の問題 | ●● | ● | ●● |
| 筋力低下/筋障害 | NR | ●● | NR |
| 筋緊張低下 | ● | ● | ●● |
| 運動障害 | ● | NR | ● |
●●● = すべて; ●● = 一般的; ● = 頻度が低い; AGAT = L-アルギニン:グリシンアミジノトランスフェラーゼ; CRTR = クレアチン・トランスポーター; DD = 発達遅延; GAMT = グアニジノ酢酸メチルトランスフェラーゼ; NR = 報告されていない
- Stockler-Ipsirogluら [2014], Khaikinら [2018]
- Stockler-Ipsirogluら [2015], DesRochesら [2016]
- van de Kampら [2013a], Bruunら [2018]
- 新生児期または幼児期から診断され、治療コンプライアンスが良好な患者は、発達のマイルストーン、認知機能、IQが正常である可能性があることに留意する。
GAMT欠損症
現在までに、GAMTの両方のアレルの病原性バリアントを持つ約130名が、同定されている [Stockler- Ipsirogluら 2014年、Khaikinら 2018]。本疾患に関連する表現型の特徴について、これらの報告に基づいて説明する。
最初の臨床症状の発症は、乳児期早期(生後3~6カ月)から2歳までである[Khaikinら 2018]。診断年齢は、新生児期から34歳までである[Stockler-Ipsirogluら 2014, Khaikinら 2018]。
最も一貫した臨床症状である発達遅延 (DD) および認知機能障害または知的障害 (ID) は、すべての罹患者に存在する。重症度は軽度から重度に及ぶ。GAMT欠損症の患者の約50%~75%が重度のDDまたはIDを有している[Mercimek-Mahmutogluら 2014, Stockler-Ipsirogluら 2014, Khaikinら 2018]。
音声言語障害:GAMT欠損症の2人の兄弟姉妹で、可変的な表現言語欠損が報告されている:発端者は、10語未満しか話さないが、妹は13歳で短い文章で話す[O'Rourkeら 2009]。
罹患者の75%以上で行動障害(多動性、自閉症、自傷行為など)が報告されている[Mercimek-Mahmutogluら 2014, Khaikinら 2018]。
GAMT欠損症における3番目に一貫した症状のてんかん発作は、罹患者の70%以上で観察される。発作のタイプには、ミオクロニー発作、全般性強直間代性発作、部分複合発作、頭部頷き発作、および脱力発作が含まれる。発作の重症度は、時々起こる発作から、次のような発作に反応しない発作まで多岐にわたる。様々な抗痙攣薬がある[Mercimek-Mahmutogluら 2014, Stockler-Ipsirogluら 2014, Khaikinら 2018]。
運動障害は、約30%の人に認められ、主にコレア、アテトーシス、ジストニア、または運動失調である[Mercimek-Mahmutogluら 2014, Stockler-Ipsirogluら 2014, Khaikinら 2018]。脳MRIにおける基底核の病理学的信号強度は、運動障害の有無にかかわらず観察される[Mercimek-Mahmutogluら 2014, Stockler-Ipsirogluら 2014, Khaikinら 2018]。発症は、通常12歳以前であるが、GAMT欠損症の若い女性が17歳で運動障害(弾道運動やジストニック運動を含む)を発症したことが報告されている[O'Rourkeら 2009]。
AGAT 欠損症
現在までに、GATMの両方のアレルの病原性バリアントを持つ16名が同定されている [Stockler-Ipsirogluら 2015年]。以下、本疾患に関連する表現型の特徴について、これらの報告に基づいて説明する。
最も一貫した臨床症状である、DDと認知機能障害またはIDは、すべての罹患者に存在する。知的障害の程度は、軽度から重度まであるが、80%以上の人が、軽度から中度の知的障害を有する。
罹患者の約10%に観察される単発の発作は、発熱の有無にかかわらず発生すると報告されている。
運動障害は、罹患者では報告されていない。
行動障害は25%に認められた。
筋力低下/ミオパチーが、50%の罹患者に認められた。
CRTR欠損症 ‒ 罹患男性
現在までに、約130名のSLC6A8の病原性バリアントが同定されている [van de Kampら 2013a、Bruunら 2018、Bahlら 2020]。以下、本疾患に関連する表現型の特徴について、これらの報告に基づいて説明する。
最初の臨床症状の発症は、4カ月から54カ月に及ぶ[Bruunら 2018]。診断時の年齢は、1歳から66歳であり、平均寿命は、正常である可能性があることを示している。この障害が合理的に説明され、診断検査がより広く利用できるようになった現在、診断は主に、生後3年以内に行われると予想される。
DDと認知機能障害またはIDは、軽度から重度までのすべての罹患した男性患者に認められた。罹患男性の85%が、4歳まで軽度から中度のIDを持ち、18歳以上の罹患男性の75%が、重度のIDを持っていた[van de Kampら 2013a]。成人の1人は、進行性の認知機能障害であった[Kleefstraら 2005]。
音声言語障害:音声の発達は、すべての罹患男性で遅れていた。最初の言葉は、平均3.1歳であった(年齢範囲:9か月から10歳)。10歳以上の男性では、14%が音声の発達がなく、55%が単語を話すことができ、31%が文を話すことができた[van de Kampら 2013a]。
オランダの血縁関係のない2家族から罹患した4人の男児の神経心理学的プロファイルでは、意味語用法の言語障害(言葉の意味を理解することが困難)に口腔運動障害が見られた[Manciniら 2005]。
てんかん発作は、罹患した男性患者の59%に見られた。最も一般的な発作のタイプは、全般性強直間代発作と単純または複雑な部分発作で、二次的な全般化を伴うものと伴わないものがあった。欠神発作とミオクロニー発作はまれであった。発作の発症は、1歳から21歳の間であった[van de Kampら 2013b]。難治性てんかんの報告は、10人未満であった[Mercimek-Mahmutogluら 2010, van de Kampら 2013a]。
運動障害:広範な歩行または運動失調と、ジストニアまたはアテトーゼが、罹患男性のそれぞれ29%と11%に報告された[van de Kampら 2013a]。
行動障害:罹患男性の 85%で報告された。最も一般的な行動障害は、注意欠陥および/または多動性 (55%) および自閉的特徴 (41%) であった。その他の行動障害としては、社会不安や内気 (20%)、定型的行動 (20%)、衝動的行動 (27%)、攻撃的行動 (19%)、自傷的行動 (10%)、強迫的行動 (8%) が報告された[van de Kampら 2013a]。
その他の神経学的臨床的特徴:筋緊張低下は、男性の40%にみられた。痙縮は、男性の26%に認められた。4人に、軽度(感音性)難聴があった。9人の罹患男性に、斜視または両側外転神経麻痺がみられたと報告されている。ミオパチー様顔貌、眼瞼下垂、関節弛緩(おそらく筋緊張低下による二次的なもの)、筋量減少も報告されている[van de Kampら 2013a]。
その他の非神経学的臨床的特徴
- 特徴的な形態変化;小頭症、広い額、中顔面後退、高い口蓋、短い鼻、顕著な鼻梁、耳の違い(折り込み不十分の耳介、大きな耳、および/または陥没耳)、落ちくぼんだ目、第5指短指、および細身の体格など。45%の罹患男性に報告された[van de Kampら 2013a, van de Kampら 2013b]。
- 消化器系の所見として、体重増加不良、嘔吐、便秘、イレウス(便秘に続発すると考えられる)、肝炎、胃・十二指腸潰瘍、食道ヘルニア(CRTR欠損と関連があるかどうかは不明)が罹患男性の35%で報告されている[van de Kampら 2013a]。
- 心臓の特徴:1名の罹患男性に、QT延長障害がみられた[van de Kampら 2013a]。CRTR欠損症の男性7名 (39%) は、心電図でQTcの延長を認めた。これらの患者はまた、心エコーで左心室内径(拡張期)の増大と左心室後壁径(拡張期)の減少を示した[Levinら 2021]。
- 成人期の医学的懸念:罹患男性101人中21人が成人(年齢18歳以上)であった。筋萎縮性顔貌、眼瞼下垂、外眼筋麻痺、またはパーキンソニズムを呈した。一部の成人では、巨大結腸、イレウス、腸管穿孔、胃・十二指腸潰瘍に至る慢性便秘が報告されている[van de Kampら 2013a]。
CRTR欠損症 ‒ ヘテロ接合性の女性
家族特異的なSLC6A8病原性バリアントを、ヘテロ接合で持つ女性は、一般的に無症候性または軽度のIDである [van de Kampら 2011]。病原性バリアント対立遺伝子に有利なX染色体不活性化の歪みと、臨床表現型の重症度との間に、臨床的な相関はなかった。知的能力と、脳内1H-MRSの脳内クレアチンレベルとの間に、有意な統計的相関は認められなかった [van de Kampら 2011]。軽度の知的障害、難治性てんかん、行動障害を持つ女性(男性に類似した表現型)は、末梢血細胞におけるX染色体の偏った不活性化の証拠を持たず、脳における組織特異的なX染色体の偏った不活性化により、彼女の重度の神経学的所見を説明できた [Mercimek- Mahmutogluら 2010]。
遺伝子型と表現型の相関関係
いずれのCDDも、遺伝子型と表現型の相関関係は確認されていない。
有病率
GAMT欠損症:世界で約130人が、GAMT欠損症と診断されている。
一般人口におけるGAMT欠損症の推定発生率は、2,640,000分の1から250,000分の1である[Desrochesら 2015, Mercimek-Mahmutogluら 2016]。これは、1,500,000人以上の新生児を、スクリーニングしたGAMT欠損症の、試験的新生児スクリーニングプログラムからの情報と一致しており、現在までに、新生児のうち2人が、GAMT欠損症と確定診断されている[Mercimek-Mahmutogluら 2012, Pasqualiら 2014, Pittら 2014, Stockler-Ipsirogluら 2014, Sinclairら 2016, Hartら 2021]。
ユタ州とニューヨーク州の、新生児集団におけるGAMT欠損症の推定発生率は、1:405,655であった [Hartら 2021]。
神経疾患または重度の知的障害を持つ患者の小規模な研究では、GAMT欠損症は、1.1%存在することがわかった [Cheillanら 2012]。
AGAT欠損症:AGAT欠損症の推定保因者頻度は、ExAC Browser Betaデータベースを用いて、一般人口の1,292人に1人 (0.077%; CI=0.06%-0.10%) であった[DesRochesら 2016]。
CRTR欠損症:CRTR欠損症は、家族性または非家族性の、IDを持つ49人から4,426人に及ぶ、多くのコホートで研究されている。これらの研究は、van de Kampら [2014] によってまとめられ、IDを持つ男性における有病率は、0.4%~1.4%と推定された。最近の研究では、クレアチントランスポーター欠損症の有病率は、神経発達障害を持つ患者で2.64%であった[Bahlら 2020]。
遺伝的に関連する(対立遺伝子)疾患
このGeneReviewで議論されている以外の表現型は、GAMT、GATM、SLC6A8の生殖細胞系列病原性バリアントとの関連性は知られていない。
鑑別診断
1H-MRSで検出された脳の部分的クレアチン欠乏症で、尿、血漿、CSF中のグアニジノ酢酸 (GAA) 濃度が正常で、尿中のクレアチン/クレアチニン比が正常な人は、表3にまとめた障害を考慮する必要がある。
表 3. クレアチン欠乏症の鑑別診断で注目される疾患
| 遺伝子 | 障害 | MOI | 生化学的特徴 | 臨床的特徴 |
|---|---|---|---|---|
| ALDH18A1 | P5CS欠損症 1 | AR AD | 二次的(脳)クレアチン欠乏症 | 異形症、DD、痙性、筋障害、成長遅延 |
| ASL | アルギニノコハク酸リアーゼ欠損症 | AR | 二次性(脳)クレアチン欠乏症2; ↑血漿クレアチンレベル3 | DD、痙攣、運動失調、結節性裂毛症、高アンモニア性脳症、成長遅延、肝繊維症、肝硬変 |
| ASS1 | アルギニノコハク酸シンテターゼ欠損症 (citrullinemia type I) | AR | 二次的(脳)クレアチン欠乏症 2; 著しく ↓ 血漿クレアチン濃度 3 | DD、痙攣、運動失調、高アンモニア性脳症、成長遅延、肝硬変 |
| OAT | オルニチンアミノトランスフェラーゼ欠損症(脈絡膜と網膜の脳回様萎縮) (OMIM 258870) | AR | 二次的(脳)クレアチン欠乏症4 | 進行性脈絡網膜変性および周辺視野の喪失、近視、夜盲症、後嚢下白内障、近位筋力低下 |
| OTC | オルニチントランスカルバミラーゼ欠損症 | XL | 著しく ↓ 血漿クレアチン 3 | DD、発作、運動失調、高アンモニア性脳症、成長遅延 |
| PC | ピルビン酸カルボキシラーゼ欠損症 | AR | ↓ 脳内クレアチン 1H-MRS 5 | DD、発作、痙性、筋緊張低下、肝腫大、乳酸アシドーシス |
| SLC25A15 | 高アンモニア血症、 高オルニチン血症、 ホモシトルリン尿症症候群 | AR | 著しく ↓ 血漿クレアチン濃度 3 | DD、けいれん、痙性、筋緊張低下、成長遅延、肝腫大 |
| SLC7A7 | Lysinuricタンパク質不耐性 | AR | ↑ 血漿クレアチンレベル 3 | 低身長、嘔吐、成長遅延、呼吸不全、肝腫大、下痢、骨粗鬆症 |
1H-MRS = プロトン磁気共鳴分光法; AD = 常染色体顕性(優性); AR = 常染色体潜性(劣性)遺伝; DD = 発達遅滞; MOI = 継承様式; P5CS = デルタ-1-ピロリン-5-カルボン酸シンセターゼ; XL = X連鎖
臨床的マネジメント
クレアチン欠乏性 (CDD) の診療ガイドラインは発表されていない。いくつかの文献では、GAMTおよびCRTR欠損症に対する管理の推奨事項が記載されている[Stockler-Ipsirogluら 2014、Bruunら 2018、Khaikinら 2018]。
初期診断後の評価
CDDと診断された人の疾患の程度やニーズを把握するために、表4にまとめた評価(診断に至った評価の一部として実施されていない場合)が推奨される。
表4. クレアチン欠乏症の初回診断後に推奨される評価方法
| システム/懸念 | 評価 | コメント |
|---|---|---|
| 神経学的 | 神経学的評価 |
|
| 発達 | 発達/神経心理学的評価 |
|
| 運動障害1 |
|
以下の評価を含む:
|
| 行動学的 | 神経行動学的評価 | 12ヶ月を超える年齢を対象としたスクリーニング検査:
|
| 治療に関連する腎症の可能性 | ベースライン腎機能研究 |
|
| 心臓の懸念2 |
|
|
| 成長不良 | 栄養士による摂食と成長の評価 | 吸引力がある場合は、評価を検討する。 |
| 遺伝カウンセリング | 遺伝学の専門家による3 | 罹患した患者と家族に、特定CDDの性質、MOI、含意を知らせ、医療的・個人的な意思決定を促進すること。 |
| 家族のサポートとリソース | 以下の必要性を評価する
|
必要に応じたソーシャルワークのサポート |
ADHD = 注意欠陥/多動性障害; ADL = 日常生活動作; ASD = 自閉症スペクトラム障害; CDD = クレアチン欠乏症; eGFR = 推定糸球体ろ過量; GFR = 糸球体ろ過量; MOI = 継承様式; OT= 作業療法; PT = 理学療法
- GATM欠損症では一般的に存在しない
- CRTR欠乏症の方の考慮事項
- 医療遺伝専門医、認定遺伝カウンセラー、認定上級遺伝看護師
症状に対する対応
原因を問わない一般的なCDDの扱いを表5に示す。
CDDのタイプに応じた医療処置は、表6 (GAMT欠損症)、表7 (AGAT欠損症)、表8 (CRTR欠損症) に示すとおりである。
表5. クレアチン欠乏症患者の症状に対する治療法
| 症状/懸念 | 治療 | 考慮事項/その他 |
|---|---|---|
| 発達遅滞/知的障害と行動の問題 | 発達遅滞/知的障害の管理問題を参照。 | |
| てんかん | 経験豊富な神経内科医による抗てんかん薬による標準的な治療法 |
|
| 運動障害 | 運動障害専門医による治療 |
|
発達遅延 / 知的障害マネッジメントの課題
以下の情報は、米国における発達遅延/知的障害者に対する典型的な管理推奨事項であり、標準的な推奨事項は国によって異なる場合がある。
0 ~ 3歳:作業療法、理学療法、言語療法、摂食療法、乳児精神保健サービス、特殊教育者、感覚障害専門家などを利用するために早期介入プログラムへの紹介が推奨される。米国では、早期介入は連邦政府が資金提供するプログラムで、すべての州で利用でき、個々の治療のニーズに合わせて家庭内サービスを提供している。
3 ~ 5歳:米国では、地元の公立学校区を通じた発達段階の就学前教育が推奨されている。また、運動、言語、社会、認知の遅れが認められる場合は、個別教育計画 (IEP) が策定される。早期介入プログラムは通常、この移行を支援する。医学的に不安定で通園できない子供には、在宅サービスが提供される。
全年齢対象:適切な地域、州、教育機関の関与を確保するために、発達小児科医と相談することが推奨される(米国)。また、保護者が生活の質を最大限に高めることができるよう支援する。考慮すべきいくつかの問題を、以下に記す。
- IEPサービス:
- IEPは、資格のある子供たちに、特別に設計された指導と関連サービスを提供するものである。
- IEPのサービスは毎年見直され、変更が必要であるかどうか判断される。
- 特別支援教育法では、IEPに参加する児童は、学校で実現可能な最も制限の少ない環境に置かれ、適切な時に適切な場所で、できる限り一般教育に参加することが義務付けられている。
- PT、OT、言語サービスは、その必要性が、子供の学問的教材へのアクセスに影響する範囲内で、IEPの中で提供されることになる。それ以後は、罹患者のニーズに基づいた民間の支援療法が検討されることがある。治療の種類に関する具体的な推奨は、発達小児科医が行うことができる。
- 子供が10代になると、移行計画について話し合い、IEPに組み込む必要がある。IEPのサービスを受けている場合、公立学校区は、21 歳までサービスを提供することが義務付けられ ている。
- 504プラン(Section 504:障害に基づく差別を禁止する米国連邦法)は、クラスの前方座席、支援技術装置、教室の書記、授業間の延長時間、課題の変更、テキストの拡大などの便宜や修正を必要とする人のために検討される。
- 発達障害者福祉局 (DDA) への登録が推奨される。DDAは米国の公的機関で、有資格者にサービスやサポートを提供する。適格性は州によって異なるが、通例では診断名および/または関連する認知/適応障害によって決定される。
- また、収入や資源に限りのある家庭は、障害を持つ子供のために、補足的なセキュリティ収入 (SSI) の資格を得られる場合がある。
運動機能障害
総運動機能障害
- 運動能力を最大限に高め、後に発症する整形外科的合併症(拘縮、脊柱側弯症、股関節脱臼など)のリスクを軽減するために、理学療法が推奨される。
- 必要に応じて、耐久性のある医療機器や体位変換装置の使用を検討する(例:車椅子、歩行器、浴用椅子、装具、適応型ベビーカー)。
- 過緊張やジストニアなどの筋緊張異常に対しては、バクロフェン、チザニジン、ボトックス®、抗パーキンソン薬、または整形外科的処置の管理を支援するために、適切な専門家の関与を検討する。
微細な運動機能障害:摂食、身だしなみ、着替え、筆記などの適応機能に影響を与える微細な運動能力の障害には、作業療法が推奨される。
口腔運動機能障害は毎回評価し、哺乳時の窒息/嚥下、体重増加不良、頻繁な呼吸器疾患、または他に説明がつかない哺乳拒否に対しては、臨床摂食評価および/またはX線嚥下検査を受ける必要がある。子供が口から食べても安全であると仮定すると、協調性や感覚に関連した摂食の問題を改善するために、摂食治療(一般的に作業療法士または言語療法士による)が推奨される。安全性を高めるために、給餌を濃くしたり、冷やしたりすることがある。摂食機能障害が重度の場合は、NGチューブやGチューブが必要になることがある。
コミュニケーションの問題:表出性言語障害を持つ個人に対して、代替コミュニケーション手段(例えば、拡張代替コミュニケーション [AAC])の評価を検討する。AACの評価は、その分野の専門家である言語聴覚士が行うことができる。この評価では、認知能力と感覚障害を考慮して、最も適切なコミュニケーションの形態を決定する。AAC機器には、画像交換コミュニケーションのようなローテクなものから、音声生成機器のようなハイテクなものまで、さまざまなものがある。一般に考えられているのとは異なり、AAC機器は言葉の発達を妨げるものではなく、むしろ最適な言語と言葉の発達をサポートするものである。
社会的/行動的懸念
子どもたちは、応用行動分析 (ABA) を含む自閉症スペクトラムの治療で用いられる介入を受ける資格があり、その恩恵を受けることができるかもしれない。ABA療法は、個々の子どもの行動的、社会的、適応的な長所と短所を対象とし、通常、認定行動分析官と1対1で行われる。
発達小児科医との相談は、適切な行動管理戦略を保護者に指導したり、必要に応じて注意欠陥/多動性障害の治療に使用される薬などの処方薬を、提供するのに役立つ場合がある。
深刻な攻撃的行動や破壊的行動についての懸念は、小児精神科医が対応することができる。
表 6. GAMT欠損症患者の症状に対する治療法
| 症状/懸念 | 治療 | 考慮事項/その他 |
|---|---|---|
| 脳内クレアチンレベルの低下 |
|
GAMT欠乏症の症状のある場合: 1
|
| 神経毒性レベルのGAAの蓄積 |
|
|
|
bw = body weight; GAA = グアニジノ酢酸塩
- Stockler-Ipsirogluら [2014], Khaikinら [2018]<
- Stockler-Ipsirogluら [2014
- アルギニン制限の理解、食事表示の読み方、アルギニン摂取量の計算が難しいため(特にアルギニン含有量は必ずしも表示されていないため)、多くの施設では、代わりにタンパク質制限を採用している。
- 利用可能なデータベース(例:米国農務省National Nutrient Database)を使用して、食品の正確なアルギニン含有量を決定し、GAMT欠損症患者の1日のアルギニン摂取量を正確に計算することが可能である。
表7. AGAT欠損症患者の症状に対する治療法
| 症状/懸念 | 治療 | 考慮事項/その他 |
|---|---|---|
| 脳クレアチンレベルの低下 |
|
AGAT欠乏症の症状がある場合:
|
bw = 体重; ID = 知的障害
- Stockler-Ipsirogluら [2015]
- Stockler-Ipsirogluら [2015]
- Battiniら [2006], Stockler-Ipsirogluら [2015], Battiniら [2017]
表 8. CRTR欠損症の方の症状に対する治療法
bw = 体重
| 症状/懸念 | 治療 | 考慮事項/その他 |
|---|---|---|
| 脳クレアチンレベルの低下 |
|
|
1. Mercimek-Mahmutogluら [2010], Valayannopoulosら [2012], van de Kampら [2012], van de Kampら [2014], Bruunら [2018]
一次発現の予防
マニフェストの扱いを参照する。
定点調査
CDDに関する定点調査の推奨事項を、表9にまとめている。
| システム/懸念 | 評価 | コメント |
|---|---|---|
| 発達 | 発達の度合いと教育の必要性を監視する。 | 都度、対応する |
| 神経性 | 臨床的に必要とされる発作のモニタリング。 | |
| 発作、運動障害、行動問題などの新しい症状の評価。 | ||
| システム/懸念 | 評価 | コメント |
| 脳内クレアチンレベル低下治療中の場合 | 体内 1H-MRSによる脳内クレアチンレベルの測定 |
|
| GAMT、AGAT、CRTR欠乏症におけるクレアチン関連腎症の可能性を検出するために、クレアチン補充療法中の腎機能 (GFR) を評価する。 | 毎年 | |
GAMT欠損症については、以下を評価する:
|
3-6ヶ月ごと | |
| AGAT欠損の場合、定期的な検査は必要ない | ||
| CRTR欠乏症の場合、血漿GAA値および血漿アミノ酸を評価する。 1 | 3-6ヶ月ごと |
GAA = グアニジノアセテート; GFR = 糸球体濾過量
- アルギニンやグリシンを大量に補給すると、GAA濃度が上昇することがある。
リスクのある親族に対する評価
クレアチン欠乏症のリスクのある新生児を評価し、早期の診断と治療を可能にすることが適切である。(注:CRTR欠損症の早期治療により転帰が変わることは証明されていない)。
評価内容は、以下の通りです。
- 家系内の病原性バリアントがわかっている場合は、分子遺伝学的検査。
- 家系内の病原性バリアントが不明な場合は、生化学的遺伝学的検査。
遺伝カウンセリングを目的としたリスクのある親族の評価に関する問題については、「遺伝カウンセリング」の項を参照する。
妊産婦管理
AGAT欠損症の1名の妊娠管理が報告されている。モニタリング中の妊娠20週までに、胎児の発育と頭囲が低下した。妊婦のクレアチンレベルは低かった。妊婦の1日のクレアチン投与量を増やしたところ、出産時には正常であり、AGAT欠損症の影響はなかった [Alessandrìら 2020]。
研究中の治療法
現在、いずれのCDDも臨床試験は行われていない。いくつかの薬物療法は、CRTR欠損症の細胞株または動物モデルで研究されている。
- スイスマウス脳海馬スライスを、グアニジノプロピオン酸を用いてクレアチントランスポーターをブロックした後、ジアセチルクレアチンエチルエステル (DAC) で処理した。その結果、細胞内のクレアチンが増加し、DACはクレアチンに代謝されることがわかった。著者らは、彼らの仮説を完全に解明するために、さらなる研究が必要であると結論付けている[Adrianoら 2018]。
- Slc6a8-/yマウスに、ドデシルクレアチンエステルを、鼻腔内および脳室内に投与し、新規物体認識 (NOR) テストを適用した。
- NOR学習の回復とクレアチンの50%増加が見られた[Ullio-Gamboaら 2019]。
- 4-フェニルブチレート (4-PBA) は、SLC6A8の病原性バリアントを発現するトランスフェクトHEK293細胞において、クレアチン取り込みを増加させることが示された[El-Kasabyら 2019]。4-PBAは、CTRT活性を救済するための、ファーマコシャペロンであると考えられた。
- シクロクレアチンはCRTR欠損マウスモデルで使用され、認知機能、てんかん、行動特徴の治療に有効であった[Cacciante et al 2020]。
米国ではClinicalTrials.gov、欧州ではEU Clinical Trials Registerを検索すると、さまざまな疾患や症状に関する臨床試験情報を入手することができる。注記:この疾患に対する臨床試験は、行われていない可能性がある。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
グアニジノ酢酸メチルトランスフェラーゼ (GAMT) 欠損症とL-アルギニン:グリシンアミジノトランスフェラーゼ (AGAT) 欠損症は、常染色体潜性(劣性)遺伝することが知られている。
クレアチントランスポーター (CRTR) 欠損症は、X連鎖的に遺伝する。
常染色体潜性(劣性)遺伝 – 家族へのリスク
発端者の両親
- GAMTまたはAGAT欠損症の子供の両親は、GAMTまたはGATMの病原性バリアントを、ヘテロ接合で持っていると推定される。
- 両親とも、GAMTまたはGATM病原性バリアントに対して、ヘテロ接合であることを確認し、信頼できる再発リスク評価を可能にするために、発端者の両親に対して分子遺伝学的検査を推奨する。
- 片親のみに病原性バリアントが検出され、親の身元確認検査で生物学的母性および父性が確認された場合、発端者で確認された病原性バリアントの1つが、発端者のde novo (新生突然変異)として、または受精後にバリアントが生じたモザイク(性腺モザイク)が生じた可能性がある[Jónssonら 2017]。発端者がホモ接合性の病原性バリアント(すなわち、同じ2つの病原性バリアント)を有すると思われる場合、考慮すべき追加の可能性は以下の通りである。
- 発端者のシングルまたはマルチエクソン欠失で、配列分析で検出されず、ホモ接合の人工的な出現をもたらしたもの。
- 病原性バリアントが存在する親染色体の片親性ダイソミーにより、プローブが病原性バリアントに対してホモ接合となった場合。
- ヘテロ接合体(キャリア)は無症候性であり、本疾患を発症するリスクはない。
発端者の同胞
- 両親ともに、GAMTまたはGATMのヘテロ接合体であることが分かっている場合、罹患者の兄弟姉妹は、受胎時に25%の確率で罹患し、50%の確率で無症候性保因者となり、25%の確率で非罹患かつ非保因者であると考えられる。
- ヘテロ接合体(保因者)は無症候性で、障害を発症する危険性はない。
発端者の子
現在までのところ、GAMTまたはAGAT欠損の個体は、生殖に至らないであることが知られている。
他の家族構成員
発端者の両親の同胞は、それぞれ50%の確率で、GAMTまたはGATMの、病原性バリアントを保有していることが分かっている。
保因者の検出
リスクのある親族に対する分子遺伝学的キャリアテストは、家族内のGAMTまたはGATM病原性バリアントを、事前に同定する必要がある。
注記:GAMT及びAGAT欠損の保因者では、生化学的検査は正常であるため、保因者の同定には、家族性GAMTまたはGATM病原性バリアントに対する分子遺伝学的検査が必要である。
X-連鎖遺伝 – 家族へのリスク
男性発端者の両親
- CRTR欠損症の男性の父親は、本疾患を発症しておらず、SLC6A8遺伝子のヘミ接合体でもないため、さらなる評価または検査は必要ない。
- 複数の罹患児を持つ家系では、罹患者の母親は、絶対的ヘテロ接合体である。注記:女性が複数の罹患児を持ち、他に罹患者の親族がおらず、白血球DNAからSLC6A8病原性バリアントが検出されない場合、性腺モザイク症である可能性が最も高いと考えられる。
- 男性が唯一の罹患家族である場合、母親がヘテロ接合体であるか、罹患男性が新生突然変異でSLC6A8病原性バリアントを有するか(その場合、母親はヘテロ接合体ではない)、母親が体細胞/生殖細胞モザイク症である可能性がある[van de Kampら 2013a, van de Kampら 2014]。
- SLC6A8病原性バリアントは、85家族のレトロスペクティブ研究において、CRTR欠損症の人の30%にde novoで発生した[van de Kampら 2013a]。
- 85家族のレトロスペクティブ研究において,母親の7%が体細胞/生殖細胞モザイクを有していた[van de Kampら 2013a]。
- 母親の遺伝的状態を確認し、信頼性の高い再発リスク評価を可能にするために、母親の分子遺伝学的検査が推奨される。
- ヘテロ接合体の母親は、学習障害や軽度の知的障害、発作の既往がある場合がある [Mercimek-Mahmutogluら 2010, van de Kampら 2011] (臨床的説明、CRTR欠損症-ヘテロ接合体女性の項を参照)。
男性発端者の兄弟
兄弟姉妹へのリスクは、母親の遺伝的状態によって異なる。
- 発端者の母親が、SLC6A8病原性バリアントを持っている場合、各妊娠でそれを伝播する確率は50%である。
- 病原性バリアントを受け継いだ男性が罹患する。
- 病原性バリアントを受け継いだ女性はヘテロ接合体となり、本疾患に関連する臨床所見を発現する可能性がある(臨床的説明、CRTR欠損症-ヘテロ接合体女性参照)。
- 発端者が単発症例(すなわち、家族内で単発)であり、母親の白血球 DNA から SLC6A8 病原性バリアントが検出されない場合、母親の生殖細胞モザイクの可能性により、同胞へのリスクは低いが、一般集団より高いと推定される [van de Kampら 2011, van de Kampら 2013a]。
男性発端者の子孫
罹患した男性は、次世代に子孫を残すことが知られていない。
その他の家族
発端者の母方の叔母は、SLC6A8病原性バリアントのヘテロ接合体であるリスクがあり、叔母の子孫は性別により、病原性バリアントのヘテロ接合体(女性)またはヘミ接合体(男性)であるリスクがある。
保因者の検出
女性のヘテロ接合体を同定するための分子遺伝学的検査は、家系内のSLC6A8病原性バリアントを事前に同定することが必要である。注記:SLC6A8病原性バリアントのヘテロ接合体である女性は、本疾患に関連する臨床所見を発現することがある(臨床的説明、CRTR欠損症 - ヘテロ接合体女性参照)。
注記:ヘテロ接合体の女性は、尿中のクレアチン/クレアチニン比が正常で、脳の1H-MRSでクレアチン含有量が正常な場合があるため[van de Kampら 2011]、女性のヘテロ接合体の特定にはSLC6A8分子遺伝子検査が必要である。
遺伝カウンセリングに関連した問題
早期の診断と治療の目的のために、リスクのある親族を評価する情報については、「管理、リスクのある親族の評価」を参照する。
家族計画
- 遺伝的リスクの判定と出生前 / 着床前遺伝子検査の利用可能性について話し合う最適な時期は、妊娠前である。
- ヘテロ接合体である、あるいはヘテロ接合のリスクがある若年成人に遺伝カウンセリング(子孫への潜在的リスクと生殖の選択肢についての話し合いを含む)を行うことは適切である。
DNAバンキング:今後、検査方法や遺伝子、発症メカニズム、疾患に関する理解が進むと思われるので、分子診断が確定していない(つまり、原因となる発症メカニズムが不明な)発端者のDNAをバンクすることを検討すべきである。
出生前検査と着床前遺伝子検査
分子遺伝学的検査
GAMT、GATM、またはSLC6A8の病原性バリアントが、罹患した家族で同定されると、リスクの高い妊娠のための出生前検査および着床前遺伝子検査が可能になる。
生物学的検査
羊水中のグアニジノ酢酸 (GAA) とクレアチンを分析することにより、GAMT欠損症のリスクが高い妊娠の出生前診断が可能である。GAMT欠損症の児の母親に対し、妊娠15週目に出生前診断のための羊水穿刺を実施した。GAAは11.43μmol/L(15週目の無月経の正常範囲は2.96±0.70μmol/L)だった[Cheillanら 2006]。クレアチン欠乏症の出生前診断における生化学的検査の使用に関する情報は、非常に限られているため、分子遺伝学的検査が、出生前診断に望ましい方法である。
出生前検査の実施については、医療関係者の間や家族間で考え方の相違がある場合がある。ほとんどの施設では、出生前検査の利用は個人的な決定であると考えられているが、これらの問題について議論することは有用であろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
-
クレアチン欠乏症協会
1024 Bayside Drive
Suite 532
Newport Beach CA 92660
Email: kim@creatineinfo.org www.creatineinfo.org - MedlinePlus
- MedlinePlus
- MedlinePlus
- 米国知的障害者協会 (AAIDD)
8403 Colesville Road
Suite 900
Silver Spring MD 20910
Phone: 202-387-1968
Fax: 202-387-2193
www.aaidd.org
- 米国てんかん学会 (AES)
- てんかん財団
3540 Crain Highway Suite 675
Bowie MD 20716
Phone: 800-332-1000 (toll-free)
Email: ContactUs@efa.org www.epilepsy.com
- メタボリックサポート
UK United Kingdom
Phone: 0845 241 2173
www.metabolicsupportuk.org
- クレアチン欠乏症患者登録協会
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表 A. クレアチン欠乏症:遺伝子とデータベース
| 遺伝子 | 染色体遺伝子座 | タンパク質 | 遺伝子座固有のデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| GAMT | 19p13.3 | グアニジノ酢酸N-メチルトランスフェラーゼ | GAMT @ LOVD | GAMT | GAMT |
| GATM | 15q21.1 | グリシンアミジノトランスフェラーゼ、ミトコンドリア | GATM @ LOVD | GATM | GATM |
| SLC6A8 | Xq28 | ナトリウムおよび塩化物依存性クレアチントランスポーター 1 | SLC6A8 @ LOVD | SLC6A8 | SLC6A8 |
データは、HGNCの遺伝子、OMIMの染色体座、UniProtのタンパク質という標準的なレファレンスから編集されている。リンク先のデータベース(Locus Specific、HGMD、ClinVar)については、こちらを参照する。
表 B. クレアチン欠乏症のOMIMエントリー(OMIMで全てを見る)
| 300036 | SOLUTE CARRIER FAMILY 6 (NEUROTRANSMITTER TRANSPORTER, CREATINE), MEMBER 8; SLC6A8 |
| 300352 | 脳性クレアチン欠乏症 1; CCDS1 |
| 601240 | グアニジノ酢酸メチルトランスフェラーゼ; GAMT |
| 602360 | L-アルギニン:グリシンアミジノトランスフェラーゼ; GATM |
| 612718 | 脳性クレアチン欠乏症 3; CCDS3 |
| 612736 | 脳性クレアチン欠乏症 2; CCDS2 |
分子病因
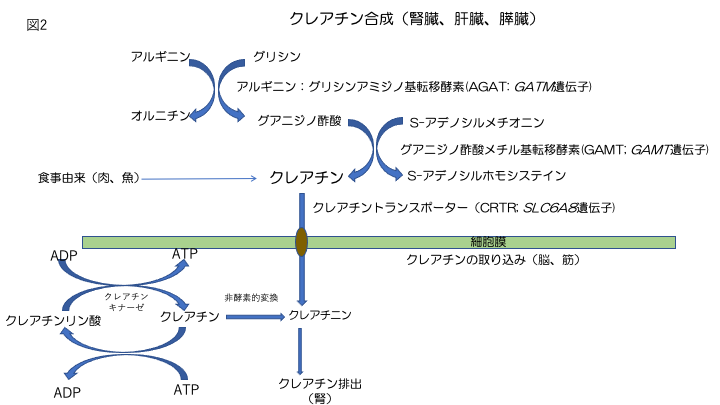
クレアチンは、2つの酵素反応によって合成される。
- L-アルギニン:グリシンアミジノトランスフェラーゼ(別名:グリシンアミジノトランスフェラーゼ、ミトコンドリア、AGAT、またはGATM)により触媒される、アルギニンからグリシンへのアミジノ基の転移により、グアニジノ酢酸(GAA)を生成する。
- S-アデノシル-L-メチオニン:N-グアニジノ酢酸メチルトランスフェラーゼ(別名:グアニジノ酢酸N-メチルトランスフェラーゼまたはGAMT)によるGAA分子のアミジノ基のメチル化。
クレアチンは、主にAGAT活性の高い腎臓と膵臓、GAMT活性の高い肝臓で合成される。脳でも遺伝子と酵素の両方が検出されている[Braissant & Henry 2008]。
合成されたクレアチンは、血液を介して利用器官(主に筋肉と脳)に運ばれ、ナトリウムと塩化物依存性のクレアチントランスポーター1(SLC6A8タンパク質)を介して取り込まれる(図2)[Wyss & Kaddurah-Daouk 2000]。このタンパク質は、骨格筋と腎臓に主に発現しているが、脳、心臓、結腸、精巣、前立腺にも存在する。クレアチン-ホスホクレアチンシャトルは、骨格筋と心筋へのエネルギー供給維持に重要な機能を有している。筋肉細胞は、クレアチンを合成せず、ナトリウム依存性の特別な輸送体であるクレアチントランスポーターを介して取り込む。
疾病の因果関係メカニズム
- GAMTまたはGATMのいずれかのバイレリック病原性バリアントは、関連する酵素の欠損をもたらし、その結果、クレアチンの合成が欠損することになる。
- SLC6A8の半接合型(またはヘテロ接合型)病原性バリアントは、クレアチントランスポーター蛋白の合成不全をもたらし、その結果、脳および筋肉へのクレアチンの輸送不全を引き起こす。
遺伝子特異的な実験室の考慮事項:SLCA8。この遺伝子の重複により生じたパラロガスコピー (paralogouscopy)が16番染色体に局在しており、遺伝子検査に支障をきたす可能性がある。
更新履歴:
- GeneReviews著者: Saadet Mercimek-Andrews, MD, PhD, FCCMG, FRCPC,Gajja S Salomons, PhD
日本語訳者:和田 敬仁 (京都大学大学院医学研究科ゲノム医療学講座)
GeneReviews最終更新日: 2022.2.10. 日本語訳最終更新日: 2022.3.14.[ in present]
![]()
