シスチノーシス(シスチン症)
(Cystinosis)
Gene Reviews著者: Galina Nesterova, MD and William A Gahl, MD, PhD.
日本語訳者: 岡村匡史、大熊喜彰、赤平百絵(国立国際医療研究センター)
Gene Reviews 最終更新日: 2016.10.6.日本語訳最終更新日: 2017.2.3.
原文 Cystinosis
要約
疾患の特徴
スチノーシスは下記の3つのタイプに分類される:
- 未治療の小児腎障害型シスチノーシスは、腎ファンコニ症候群、成長障害、低リン酸血症性/カルシウム欠乏性クル病、腎不全を引き起こす糸球体機能障害、および蓄積したシスチンによる全身性の組織・臓器の細胞機能不全を呈する。未治療の患児の典型的な特徴は、低身長、クル病および羞明(photophobia)である。体重増加不良は生後6ヶ月齢頃に気づくことが多く、腎尿細管性ファンコニ症候群(多尿、多飲、脱水及びアシドーシス)は、早ければ生後6ヶ月頃に現れる。角膜の結晶は、1歳前に現れることがあるが、通常は16ヶ月過ぎである。腎移植やシスチン除去治療をしない場合、腎型シスチノーシス患児の寿命は10才未満で、これらの介入を行えば、少なくとも40才半ばから50才くらいまで、満足のいくQOLを維持することができる。
- 中間型シスチノーシスは、発症年齢が遅いのを除き、腎型シスチノーシスと同様の症状を呈する。すべての未治療の罹患者が、通常15-25才の間に腎不全を発症する。
- 非腎型(眼型)シスチノーシスは、臨床的に角膜へのシスチン結晶蓄積による羞明のみを呈する。
診断・検査
シスチノーシスの診断は、発端者に下記のいずれか一つの所見があった場合:
- 細隙灯顕微鏡(スリットランプ)検査による角膜のシスチン結晶の同定
- 多核白血球中のシスチン含有量上昇の同定
- 培養線維芽細胞、あるいは出生時の胎盤におけるシスチン含有量の増加
- CTNS遺伝子の両アリルに病因変異を同定(分子遺伝学的検査)
臨床的マネジメント
症状の治療: 腎ファンコニ症候群は、尿細管からの電解質、重炭酸塩、無機質(ミネラル)、およびその他低分子栄養素の喪失を補充することにより治療する。小児に対しては、水分を自由に摂取でき、トイレにも行きやすい状況にする。血液をアルカリ化するため、クエン酸を補給する。クル病の予防、治療にはリン酸塩やビタミンDを補給する。骨格変形は、整形外科医と協力し、早期に対処すべきである。脱水症状が見られる期間は、補液と栄養素を補給する。腎糸球体疾患に対しては、システアミンを服用することで、細胞内のシスチンを減少させる。腎移植は最終的な治療である。システアミン点眼薬は羞明を軽減する。乳児期の体重増加不良の影響を最小限にするために、栄養は十分与えなければならない。成長ホルモン治療、甲状腺機能低下症治療のためのL-サイロキシン、糖尿病治療のためのインスリン、男性における性腺機能低下症のテストステロン補充療法は、これらすべて有益である。理学療法や言語療法は、高齢者の筋力低下や嚥下困難に有効である。
初期症状の予防: シスチン除去薬による治療は診断後できるだけ早く、あるいはもし可能であれば、出生後まもなく始めることにより、糸球体障害の進行を遅らせる。診断時にすでに腎障害がある場合は、症状を軽減することはできない。適切な対処療法とシスチン除去治療を施すことで正常な速度で成長するが、成長ホルモン療法をしなければ、一般的に低身長は回復しない。
二次的合併症の予防: 腎移植を行った後は、免疫不全や感染症の兆候をモニターする。移植前の患者では、カルニチンの補充により筋力低下を改善する。プロトンポンプ阻害剤は、システアミンによる胃酸過多を軽減する。
サーベイランス:
- 腎障害の重症度に応じて、3-6ヶ月ごとの腎臓専門医による腎機能評価
- 1-2年ごとの眼科検診
- 疾患経過をとおした骨の石灰化の評価
- 2-3年ごとの空腹時血糖値、血中テストステロン測定(男性は思春期前に始める)
- 多職種医療チームによる遅発性合併症のモニタリング
回避すべき薬物や環境:脱水、羞明がある場合には直射日光
リスクの高い親族の評価:生化学的検査と分子遺伝学的検査(発端者の遺伝子情報がわかっている場合)の両方、またはいずれか一方により、早期診断と治療が可能である。
遺伝カウンセリング
シスチノーシスは、常染色体劣性形式により遺伝する。受精段階で罹患者の同胞が発症する確率は25%、発症せずに保因者となる確率は50%、発症もせず保因者ともならない確率は25%である。リスクの高い血縁者の保因者診断とリスクの高い妊娠の出生前診断は、家族内で病気を引き起こす遺伝子変異が同定されていれば可能である。腎障害型シスチノーシスの可能性が高い妊娠においては、絨毛膜絨毛と羊膜細胞内のシスチン濃度増加に基づいた、生化学的な出生前診断が可能である。
診断
国際的なシスチノーシスの専門家が作成・承認した"Cystinosis Standards of Care" (https://cystinosis.org/images/family-support/resources/CRN_Standards_12pgloRes.pdf)がオンラインで利用可能である。
シスチノーシスを疑う所見
腎障害型シスチノーシスは、乳児および幼児に下記の臨床的所見、検査所見およびレントゲン所見がある場合に疑うべきである。
臨床的所見
- 生後6ヶ月以降の体重増加不良および成長遅延
- 嘔吐、摂食困難
- 重度の多尿、多飲および脱水
- 進行性くる病による骨格の変化;歩行可能年齢での歩行困難
- テタニー
検査所見
- 低クロール血性代謝性アシドーシス
- 腎ファンコニ症候群:電解質(ナトリウム、カリウム、重炭酸塩)、無機質(カルシウム、リン酸塩、マグネシウム)、ブドウ糖、アミノ酸、ß2-ミクログロブリンを含む尿細管タンパク質の尿からの排泄増加
- 血清アルカリフォスファターゼの上昇、低カルシウム血症、低リン血症、低カリウム血症
レントゲン所見
- 単純X線像でのくる病変化;下脚の弓状変形、骨幹端の拡大、骨端線の拡大、不正、毛ばだち(フレアやカッピング);全身性の骨減少症
- 腎超音波検査で検出される髄質性腎石灰化症、あるいは高エコー輝度
中間型シスチノーシス(若年型/遅発型)は、腎ファンコニ症候群に加え、末期腎不全を引き起こす進行性の慢性糸球体機能不全が見られた場合に疑うべきである。
眼型(非腎型)シスチノーシスは成人に羞明と、細隙灯検査において角膜のシスチン結晶の両方、あるいはいずれか一方が見られた場合に疑うべきである(図1b)。
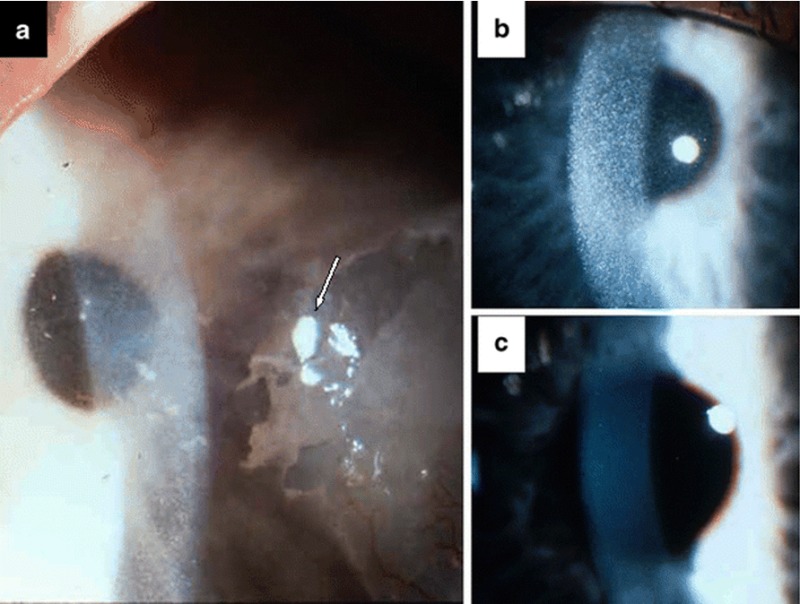
図1.シスチノーシス患者角膜の細隙灯顕微鏡(スリットランプ)検査所見
- システアミンを点眼している33歳シスチノーシス患者の帯状角膜変性症へのシステアミンの点眼は、角膜のシスチン結晶を溶解させるが、石灰化には効果がない。
- 43ヶ月齢未治療児の角膜のシスチン結晶
- システアミン点眼治療を12ヶ月行ったパネルbと同一患児
確定診断
シスチノーシスの診断は、発端者に下記のいずれか一つの所見があった場合:
- 細隙灯顕微鏡(スリットランプ)検査による角膜のシスチン結晶の同定(生後12ヶ月以降で観察できるが、通常は16ヶ月過ぎである)(図1b)
- 多核白血球中のシスチン含有量上昇
- 培養線維芽細胞、あるいは出生時の胎盤におけるシスチン含有量の増加
- 分子遺伝学的検査によりCTNS遺伝子の両アリルに病因変異を同定(表1参照)
増加した多核白血球中シスチン濃度の同定は、質量分析装置を用いるのが最もよい測定法である。
- 腎障害型シスチノーシス患者のシスチン濃度は、一般的に3.0-23.0 nnmol half-cystin/mg proteinである。
- 非腎障害型シスチノーシス患者は、1.0-3.0 nnmol half-cystin/mg proteinである。
- 保因者(ヘテロ)は、≤1.0 nmol half-cystine/mg proteinである。
- 正常値は、≤0.2 nmol half-cystine/mg proteinである。
注:
(1)シスチン測定用に白血球を調整する場合は、下記のことを注意しなければならない。
(a)多核白血球は正常量の50倍、一方リンパ球は5倍しかシスチンを蓄積していないので、リンパ球の数が顕著に多いと白血球中のシスチン量が低くなる。
(b)赤血球の混入によりタンパク量が増加してしまうため、タンパク質で補正したシスチン量が低くなる。これらの混入により、人為的に白血球中のシスチン量が低くなる。(2)イオン交換クロマトグラフィーなどを用いたアミノ酸測定は感度が低く、白血球中のタンパク質が少ないときには、測定値が信頼できない場合がある。
その他のシスチン濃度測定
シスチノーシスは培養した線維芽細胞中あるいは出産時の胎盤中のシスチン量の増加によっても診断できる。
分子遺伝学的検査
- 単一遺伝子検査:患者がヨーロッパ系の家系でなければ、CTNS遺伝子の配列を解読し、もし病因変異が一つ、あるいは見つからない場合はターゲット遺伝子の欠失/重複解析を引き続き行う。
北ヨーロッパ系の家系の患者では、まずCTNS遺伝子の57-kb欠失変異解析を行う。CTNS遺伝子の病因変異解析は、最初にフランス系カナダ人集団を対象にした検査パネルを用いるとよい。
- 原因遺伝子解析パネル:CTNSおよび着目したその他の遺伝子(類症鑑別参照)を含む原因遺伝子解析パネルも考慮される。
注:(1)パネルに含まれる遺伝子およびそれぞれの遺伝子検査の診断感度は、検査機関により様々である。 (2)いくつかの原因遺伝子解析パネルは、GeneReviewで議論されている条件に関連していない遺伝子を含んでいる。そのため、臨床医はどの原因遺伝子解析パネルが、検査にかかる費用も含め、原因遺伝子同定に最も適しているかを決定する必要がある。(3)パネルに用いられている方法は、シークエンス解析、欠失/重複解析、およびその他シークエンスに基づかない検査である。
- 網羅的ゲノム検査:一連の単一遺伝子検査と原因遺伝子解析パネルの両方、あるいはいずれか一方でシスチノーシスの臨床症状を持つ患者の診断が確定できない場合は、全エクソームシークエンス(WES)や全ゲノムシークエンスを考慮する(利用可能な場合)。このような検査は、これまでに考慮されなかった診断を可能にすることもある(例えば異なる遺伝子の変異、あるいは似たような臨床症状を示す疾患の原因遺伝子)。
表1. シスチノーシスで用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | この方法で検出できる病因変異2 を持つ発端者の割合 |
|---|---|---|
| CTNS | シークエンス解析3 | 56%5 |
| 標的遺伝子欠失/重複 解析4 |
44%5 |
- 染色体上の位置およびタンパク質名は、TableA. 遺伝子とデータベースの項を参照
- アリル変異に関する情報は、"分子遺伝学"の項を参照
- シークエンス解析により、病因ではない(benign)、病因ではない可能性が高い(likely benign)、不明(of uncertain significance)、病因である可能性が高い(likely pathogenic)、病因である(pathogenic)変異を検出する。病因変異は、遺伝子内の小欠失/挿入、ミスセンス変異、ナンセンス変異およびスプライス変異を含んでおり、一般的にはエクソンあるいは全遺伝子欠失/重複は同定されない。
- 標的遺伝子欠失/重複解析は、遺伝子内の欠失、あるいは重複を検出する。検出に利用される方法は、定量的PCR(quantitative PCR)、ロングレンジPCR、multiplex ligation-dependent probe amplification (MLPA)、単一エクソンの欠失あるいは重複を検出する、標的遺伝子マイクロアレイが用いられる。
- Shotelersuk et al [1998], Attard et al [1999]
検査特性
検査の感度や特異性を含む検査の特性に関しては、Clinical Utility Gene Card [Levtchenko et al 2014]を参照。
臨床像
自然経過
シスチノーシスは、腎障害型(古典的腎型、全身性)、中間型(遅発性腎障害型)および非腎障害型(眼型)の3つの型に分類される、CTNS遺伝子の病因変異によって引き起こされる対立遺伝子疾患である。
腎障害性および中間型(腎障害型)シスチノーシス
腎障害型シスチノーシスは、未治療の場合、体重増加不良、成長障害、腎尿細管性ファンコニ症候群、腎糸球体機能不全および腎以外の様々な組織・臓器合併症などを発症する。効果的なシスチン除去治療を行えば、シスチノーシスは、進行性、致死性腎臓病から治療可能な慢性多臓器疾患となり、寿命は未治療だと約10歳であるが、治療すれば50歳かそれ以上になる。
- 成長:未治療の腎障害性シスチノーシス乳児は、出生時は正常だが、初期には体重増加不良、後に成長障害が一般的に6-9ヶ月の間に認められる。腎臓からの栄養素喪失に加え、高頻度に嘔吐、食欲低下、摂食困難が見られ、それらが原因となり低栄養および重度の体重増加不良となる。典型的には、1歳のときの身長及び体重は3パーセンタイルである。後に、成長率は正常の60%になる。骨年齢は、大抵1−3年遅れる。頭囲は年齢別の正常範囲内である。
- 早期の適切な対症療法やシスチン除去治療により、患児の成長率は正常となる。過去には、しばしば身長は3パーセンタイル以下、体重は3パーセンタイルをやや上回る程度であったが、最近は、多くの罹患した乳幼児の身長は、年齢別の10-25パーセンタイルであり、両親の予測平均身長の範囲内になる。思春期前の子供に成長ホルモン投与を投与することで、成長速度が改善する。
- 腎尿細管性ファンコニ症候群:未治療の腎障害性シスチノーシス乳児は、腎尿細管性ファンコニ症候群(全般性近位尿細管機能障害)の徴候が、早くも生後6ヶ月で表れる。ファンコニ症候群は、腎尿細管からの水、電解質、重炭酸塩、リン酸、カルシウム、グルコース、カルニチン、アミノ酸、尿細管性タンパク質の再吸収不全である。未治療の患者は、重度の多尿(2-6L/日)、多飲、脱水及び低クロール血性代謝性アシドーシスとなり、生命にかかわる循環血液量減少の結果、特に消化管疾患を発症した場合は、時には入院治療が必要となる。
リン酸およびカルシウム排泄量の増加、血清アルカリフォスファターゼの上昇、骨変形により特徴付けられる低リン酸/低カルシウム血症性くる病は、歩行開始が遅れるほどの歩行時痛を引き起こす。ビタミンDやカルシウムの栄養欠乏はくる病を併発し、けいれん発作やテタニーを引き起こす場合がある。
重度の低カリウム血症は心臓の電気伝導を悪化させる。時に低ナトリウム血症や低マグネシウム血症も生じる。
くる病、テタニー、アシドーシスおよび検査所見の異常を解決するためには、腎臓からの喪失を補充する入念な治療が必要である。尿細管障害が起こっていない出生直後からシスチン除去治療を開始することで、腎尿細管性ファンコニ症候群を軽減することができる。しかしながら、通常診断される時期にはすでに腎尿細管障害があり(すなわち、~1歳)、障害は不可逆性である。
- 糸球体機能障害:未治療の腎障害性シスチノーシスの自然経過の中で、糸球体機能は徐々に悪化し、10歳頃までの腎不全となる。血清クレアチニン濃度は、5歳までは1.0mg/dLを超えないが、一度上昇すると指数関数的に上昇する。多くの患者は、著明な蛋白尿を呈し、時に顆粒円柱や顕微鏡的血尿を伴うネフローゼの域に達する。
システアミンの服用など、早期にシスチン除去治療を行えば、糸球体障害の進行は緩やか、あるいは停止し、腎移植の必要性を遅らせる、あるいはなくすことができる。
- 腎以外の合併症:適切な治療が施されない場合、骨髄、肝臓、小腸、筋肉、脳、脾臓、眼、甲状腺、膵臓および精巣など、ほとんど全ての組織・臓器にシスチンが蓄積し、いくつかの合併症が、腎移植前に起こる。
- シスチノーシスの患児は、軽度の頭蓋顔面変形、気道容積の減少、歯の発達遅延、および永久歯への生え変わりの遅れが見られる。
- 羞明は、角膜への結晶の蓄積が原因で、一般的に10代後半に発症する。
- 甲状腺機能低下症は、一般的に10代後半に発症する。
- 発汗が障害され、罹患者(児)は熱疲労になりやすい。
- 頭痛や乳頭浮腫を伴う良性頭蓋内圧亢進症。
- 思春期は、一般的に1−2年遅れ、未治療の男児は、原発性性腺機能低下症を呈する。
- 未治療の場合、男性に子供ができたことはなく、何人かの女性は健常児を出産している。
- 処方に従ったシステアミン内服治療により、男性の原発性性腺機能低下症が予防できるかは、現在のところまだわからない。
- 遅発性の異常症状:腎移植した後(~20-40歳)、システアミンを服用していない患者において、腎臓以外の臓器における長期間のシスチン結晶蓄積により、別の合併症が起こる。
- 筋肉内のシスチン量の増加により、60%の患者で空胞性ミオパチーを発症する。全身性ミオパチーは、進行性の筋萎縮と筋力低下を引き起こす(図2a, 2c, 2d)。口腔運動障害により、嚥下困難、摂食障害となる。筋電図検査は、ミオパチーパターンを示す。
- 外因性の胸筋機能不全により、肺実質外の換気制限が引き起こされ、肺機能検査のFVCやFEV1値の低下を伴う肺機能不全となる。
- 消化器症状は、逆流、運動不全、食道炎、胃/十二指腸潰瘍、門脈内圧亢進症を伴う結節性再生過形成による肝腫大、膵外分泌不全、炎症性腸疾患、腸穿孔、腹膜炎である。
- 循環器系の症状は、血管の石灰化、高コレステロール血症を伴う閉塞性動脈硬化症(図2e)、末期腎不全(ERDS)やレニン依存性高血圧、拡張性心筋症、大動脈瘤などの複合要因によって引き起こされる、動脈疾患が含まれる。これらすべての因子は心血管疾患罹患率に寄与し、心筋梗塞や神経血管インシデントのリスクを増大する。
- 代謝性骨疾患は、骨への直接的なシスチンの沈着、無機質の不均衡、腎移植前の腎性骨形成異常症の結果として発症する。
- 凝固能亢進や凝固能低下は、腎機能不全や血小板凝集能障害が原因でおこる。
- 中枢神経系の石灰化(図2f)、髄液の吸収障害による水頭症を伴う良性頭蓋内圧亢進症、大脳萎縮による中枢神経系の実質性機能の悪化が、様々な重症度の脳症を引き起こす。たまに、運動麻痺あるいは仮性球麻痺を伴う脳血管インシデントが起こる場合がある。
- 知的能力は正常下限であり、患児は大抵は平均的な学力を有している。彼らは、視覚および空間認知能力に障害があるが、言語能力および知的機能は保たれている。独特の行動や心理社会的問題点は、慢性疾患:ERDS、腎透析、長期入院、ステロイドを含む複数の治療薬による治療、に由来する。
- 遅発性眼合併症:前眼房、虹彩、毛様体、脈絡膜、眼底及び視神経におけるシスチン沈着は、下記の症状として現れる。
- 前眼部の問題点:水晶体表層前面の結晶沈着、帯状角膜変性症(図1a)、角膜周辺部の血管新生および虹彩後癒着
- 後眼部の問題点:疾患後期における視覚機能不全を引き起こす視細胞変性を伴う色素性網膜症。
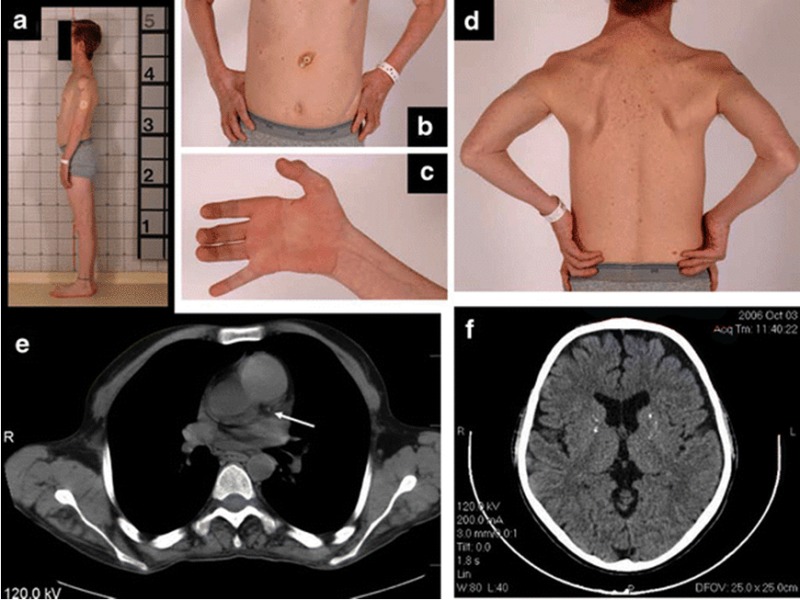
図2.
腎障害型シスチノーシスの37歳男性
- 細
- 身の体格 胃瘻管
- 母指球、小指球筋および骨間筋の萎縮による鷲手
- 上部体幹の筋肉萎縮
- 胸部CT検査:左冠状動脈の石灰化(矢印)
- 脳CT検査:大脳基底核の石灰化と萎縮
中間型シスチノーシス:腎尿細管性ファンコニ症候群、成長遅延、羞明、糸球体機能障害など初期症状はすべて腎障害型シスチノーシスと同様であるが、大部分は思春期に発症する。
病理組織診断:糸球体の電子顕微鏡像では、足突起の融合、ボウマン膜の肥厚および間質細胞内にシスチン結晶が観察される。シスチノーシスの糸球体では、巣状糸球体硬化症が見られる。
病態生理:腎尿細管性ファンコニ症候群の病態生理は、研究中である。いくつかのメカニズムが提唱されている。
- 細胞内ATPの枯渇を伴う、近位尿細管細胞内のシスチン蓄積のよるナトリウム依存性リン酸共輸送体阻害
- 腎障害型シスチノーシスでよく報告されている近位尿細管変性
- 不適切なアポトーシスの増加により、近位尿細管細胞死が進行し、尿細管消失糸球体や腎機能不全を引き起こす
- 尿細管上皮細胞におけるATP誘導性Ca2+放出感受性の増加
- 細胞シグナル変化による細胞接着不全および細胞の運動性増加
非腎型シスチノーシス
眼型シスチノーシスは、未治療だと羞明のみ感じる。
遺伝子型と臨床型の関連
一部遺伝子型と臨床型の関連がわかっている。
- 腎障害型シスチノーシス患者においては、CTNS遺伝子の57-kb欠失を含めた欠失型変異が、重症型、古典的(早期発症あるいは乳児型)シスチノーシスを引き起こす。
- (白血球中シスチン蓄積が低いなど)明らかな残存活性がある場合は、しばしばCTNS遺伝子のミスセンス変異である。中間型(遅発腎障害型)あるいは非腎型(角膜および骨髄に結晶があるものの、腎障害はない)シスチノーシス患者では、片方のアリルに腎障害型シスチノーシスを引き起こす重度の病因変異を有し、もう片方のアリルには軽度の病因変異を有している。軽度の変異は、p.Gly197Argとc.853-3C>Gである。眼型シスチノーシスの組織特異性は、組織特異的なスプライシング因子によるものかもしれない。
- p.Ser139Pheは、若年型の表現型を引き起こす病因変異の可能性がある。
- CTNS遺伝子とその隣接遺伝子を巻き込んだ欠失は、隣接遺伝子症候群になり、古典型シスチノーシスに比べ、より複雑な表現型を呈するかもしれない。例として、染色体17p13の57-kb欠失は、隣接するTRPV1まで広がっているため、末梢単核細胞におけるTRPV1遺伝子の転写の調節機能低下を引き起こす。
- ヘテロ接合体は、ライソゾーム内で50%のトランスポーター活性を有している。
- シスチノシンの活性喪失により、活性酸素シグナル、あるいはタンパク質cysteinylationを含む他のトランスポーターや経路の制御されていない活性化が引き起こされるかもしれない。シスチノーシスでは、リソソームの他の機能も障害される可能性もある。シスチノーシス患者の末梢血サンプルを用いた全ゲノム発現プロファイル解析により、腎障害性シスチノーシスに関連する修飾遺伝子や経路が同定されている。シスチノーシスの表現型を調節するこれらの遺伝子の役割を明らかにするためには、さらなる研究が必要である。
命名法
腎障害型シスチノーシスは、乳児型シスチノーシスとも呼ばれる。
中間型シスチノーシスは、若年性腎障害型シスチノーシスとも呼ばれる。
"成人型"および"良性"シスチノーシスは、命名を"眼型"および"非腎障害型"あるいは"眼非腎障害型"シスチノーシスに置き換えるべきである。
頻度
シスチノーシスは10-20万人に1人の頻度で発症し、世界中のあらゆる民族で見つかっている。フランスのブルターニュでは、2万6000千人に1の頻度である。シスチノーシスは、小児腎不全の5%をしめる。
疾患関連遺伝子
CTNS遺伝子に関連した病因変異について、このGeneReviewで取り上げた以外の表現型は知られていない。鑑別診断
腎尿細管性ファンコニー症候群
未治療の腎障害性シスチノーシスは、小児期の腎尿細管性ファンコニ症候群で、最もよく特定できる原因である。類似症状を呈する疾患は、以下の通りである。
- ウィルソン病は銅の代謝異常であり,肝障害,神経障害,精神症状およびこれらの症候が組み合わさった病態を呈する疾患で、発症時期は3歳から50歳以上である。ウィルソン病の診断は血清銅の低値,血清セルロプラスミン値の低値,カイザー-フライシャー角膜輪、そして尿中への銅の排泄量の増多加である。ATP7B遺伝子の病因変異で発症し、常染色体劣性遺伝型式である。
- ロウ(Lowe)症候群(眼脳腎症候群、OCRL)は男性にみられ、眼(白内障、緑内障、視力低下)、中枢神経(筋緊張低下、知的障害)、腎臓(ファンコニ症候群)を発症する。糸球体硬化症や腎不全は緩徐に進行し、しばしば10歳以降に気づかれる。診断は、培養皮膚線維芽細胞のPhosphatidylinositol 4,5-bisphosphate 5-phosphatase活性低下(正常の10%以下)を証明する。大部分の男性と保因者女性では、責任遺伝子であるOCRL1の病因変異が同定できる(OCRL1遺伝子変異は、Dent病の原因でもあり、Dent病は腎ファンコニ症候群の典型的な所見を示す)。遺伝様式は、X染色体劣性遺伝である。
- 古典的ガラクトース血症は、ガラクトース代謝異常であり、未治療の乳児において摂食障害、発育障害、肝細胞障害、出血および敗血症をきたす疾患である。ガラクトース-1-リン酸ウリジルトランスフェラーゼ (GALT) 酵素活性欠損が最も頻度が高い。古典的ガラクトース血症の診断は、赤血球のガラクトース-1-リン酸濃度の増加、GALT酵素活性低下およびGATL遺伝子両アリルにおける病因変異の同定の、すべてあるいはいずれか一つによって行う。遺伝様式は、常染色体劣性遺伝である。
- 糖原病I型(GSD type I)は、主に肝腫大と低血糖をきたす。
- 高チロシン血症I 型は、乳児期に重度の肝障害をきたし、有機酸分析においてチロシン代謝物の異常を示す。
- 腎尿細管性ファンコニ症候群による尿糖は、糖尿病と誤診される場合がある。
- 多尿はしばしば尿崩症と誤診される。
- 電解質の異常は、バーター症候群が疑われる。
- シスチノーシスによるくる病は、ビタミンD 欠乏性くる病と間違われる。
眼型(非腎型)シスチノーシス
多発性骨髄腫は、非腎型シスチノーシスと同様の羞明や角膜結晶を引き起こす。
臨床的マネジメント
国際的なシスチノーシスの専門家に作成・承認された、シスチノーシスの臨床診療指針が公表されている。"Cystinosis Standards of Care" を参照。
最初の診断時における評価
病気の程度やシスチノーシス罹患者のニーズを確かめるために、下記の評価法が推奨される。
最初に診断されたどの年齢においても:
- 年齢別身長・体重曲線
- 腎尿細管および糸球体機能評価、特に、血清クレアチニン、リン酸、重炭酸塩、カリウム濃度;尿中クレアチニン、リン酸、重炭酸塩、カリウム、グルコース、タンパク質濃度。尿中アミノ酸損失量を測定することは、腎ファンコニ症候群の重症度を判断する目安になる。
- 糸球体濾過率(GFR)、あるいはクレアチニンクリアランス検査
- 甲状腺機能検査
- 脂質パネル検査
- 超音波検査による腎石灰沈着の評価
- 眼科検査;細隙灯検査(角膜合併症の評価)、ERG(electroretiogram, 網膜電位図)(網膜合併症の評価)、眼底検査(頭蓋内圧亢進症の評価)
- 臨床遺伝医および遺伝カウンセラーの両方あるいはいずれか一方との相談
最初の診断時年齢が高い場合:
- 思春期前後の男性;血清テストステロン、FSH、LH測定
- 糖尿病の徴候がある場合には糖負荷試験
- 眼底検査を含む一般眼科検査
- エックス線検査およびDEXAスキャンによる骨格変形を引き起こす代謝性骨疾患の評価
- 臨床遺伝医および遺伝カウンセラーの両方あるいはいずれか一方との相談
症状の治療
腎臓内科医、代謝疾患専門医、眼科医、神経科医、消化器内科医、栄養士、心理学者を含む多職種医療チームが、シスチノーシス患者の治療にあたることが推奨される。
腎ファンコニ症候群
- 腎尿細管から喪失した電解質、重炭酸塩、ミネラル、およびその他低分子栄養素の補給。
- 小児に対しては、水分を自由に摂取でき、トイレにも行きやすい状況にする。血液をアルカリ化するため、クエン酸を補給する。
- くる病を防ぎ、治療するためのリン酸の補給;消化管からのリン酸やカルシウムの吸収を助けるための、ビタミンDを補給
- 整形外科専門医のサポートによる骨格異骨格異常の早期治療
- 脱水症状が見られる期間は、補液と栄養素の補給に注意が必要である(1日当たり2-6Lの尿と共に電解質が確実に消失する)。
腎糸球体症
- シスチン除去薬であるシステアミン服用による細胞内シスチン濃度の減少(一次病変の予防を参照)。
- 腎移植。腎代替療法は、クレアチニンクリアランスが20mL/min/1.73m2以下に低下したときや、高窒素血症および高血圧が急速に進行した際に必要となる。腎代替療法が必要になるまでの期間は、年齢に対する血清クレアチニン値の逆数が、およそ0.1に達した時点である。症状により腎移植が必要な時期が決定される。
眼科的な症状 は、症状に応じてシスチン除去薬を用いて治療する。
- 角膜のシスチン結晶蓄積により生じる羞明は、太陽光を避け、サングラスの着用し、さらに市販の目薬で眼を潤すことにより、改善することができる(一次病変の予防を参照)。
- 角膜移植は、再発性角膜潰瘍からくる難治性の痛みがある場合に、ごく稀に行われる。
- 網膜合併症は回復しない。
成長
シスチノーシスの子供の成長には、良好な栄養状態、十分なリン酸の補給、および粘り強い細胞内シスチン除去が必要である(一次病変の予防を参照):
- 栄養は成長に十分な量でなければならない。
- 成長ホルモン療法により、シスチノーシスの子供たちは、正常に追いつく成長を示し、身長も正常範囲になる。
- 早期、かつ入念なサプリメントや経口システアミンの治療により、成長ホルモン療法が不要となる。
その他
- 甲状腺機能低下症;L-チロキシン経口補充
- 糖尿病;インスリン
- 原発性性腺機能低下症(男性);二次成長を促すテストステロン
- 高齢者シスチノーシス患者の筋力低下や嚥下障害のための運動。手の腱移行術は、部分的に筋力を改善させる。
- 言語療法、理学療法
- 良性頭蓋内圧亢進症の標準的な治療。他の中枢神経性合併症は回復しない。
- 嚥下障害、低栄養、および誤嚥のリスクがある場合は、胃瘻による栄養補給(図2b)。
一次病変の予防
システアミン酒石酸塩(Cystagon®)によるシスチン除去治療により、腎障害性シスチノーシス患者の治療管理と予後が劇的に改善した。現在、国際的に広くシステアミンがシスチノーシスの治療に使用されている。フリーのチオール(SH基)が、シスチン蓄積細胞のシスチン量を90%以上減少させる。Wilmer(2011)らは、システアミンが細胞内のグルタチオンレベルを上昇させ、グルタチオンの酸化還元状態を回復することを見いだしている。
システアミン治療は、年齢および移植の有無にかかわらず、すべての患者に対して考慮されるべきである。早期、かつ入念な服薬により、多くのシスチノーシス患者は、20代まで腎移植の必要性がなく生存する。
- 規則的で入念なシステアミン治療により、末期腎不全、甲状腺機能低下症の発症を予防、あるいは遅らせ、成長を促進し、筋実質のシスチンを枯渇させる。
- 腎臓の成長と機能喪失よりも機能獲得のためには、診断後すぐにシステアミン治療を開始することがきわめて重要である。
- シスタゴン(Cystagon®)は、小児は1日60-90mg/Kg(1日1.3-1.9 g/m2)、成人は500mgを6時間毎に内服する。小児、成人共に、可能であれば、白血球中シスチン濃度(内服後、5-6時間に測定)が1.0nmol/half-cystine/mg protein以下になるようよう投与量を調整する。
- プロシスビ(Procysbi® 、システアミン酒石酸塩)は、6歳以上の患者を対象とした徐放性カプセルであり、12時間毎に服用する。プロシスビはシスタゴンと同等に、血中シスチン量をコントロールする。
- システアミン治療の副作用として、その不快な臭気や味による、吐き気(胸やけ)や嘔吐がある。システアミンは、胃酸分泌を促進するガストリン合成、および胃酸の産生を増加させる。オメプラゾール(プロトンポンプ阻害剤)と一緒に服用するとよい。
- システアミンの長期服用により、大部分のシスチノーシス遅発性合併症を回避することができる。
- 処方どおりシステアミンを内服したとしても、システアミン塩酸塩点眼薬、あるいはシストラン™ (システアミン点眼液)0.44%が、角膜の結晶を溶解させるためには必要である。システアミン点眼薬0.55%溶液(保存薬として塩化ベンザルコニウム0.01%含有)を、1日10-12回点眼する。コンプライアンスが良好であれば、数週間以内に羞明の症状が軽減する(図1b、1c)。全身性のシステアミン治療により、網膜合併症を寛解、あるいは発症を遅らせる。
二次病変の予防
腎移植を受けた患者においては、免疫不全や感染症の兆候をモニターすべきである。
腎移植前の患者にカルニチンを補充することで、筋力が回復する場合がある。
システアミン誘導性胃酸分泌亢進や消化管症状は、オメプラゾールのようなプロトンポンプ阻害剤の服用により改善する。
サーベーランス
腎障害性シスチノーシスの患者は、病気の重症度に基づいて臨床検査がなされるべきであり、それら検査には腎機能、内分泌、眼科、神経および心機能検査も含まれる。
- 腎臓専門医による3-6ヶ月毎(腎機能低下の重症度に依存)の腎機能評価。
- 症状が安定している場合、少なくとも3-6ヶ月毎の、腎機能検査、電解質、甲状腺機能検査。
- カルシウム、リン酸、アルカリフォスファターゼ、副甲状腺ホルモンの血清濃度。単純X線検査はDEXAスキャンと同様に、骨折の予測素因になる骨減少症や骨脆弱性を検出する。それらを診断確定後すぐ開始して、継続的に実施する。
- 頭蓋内圧の上昇のスクリーニングのため、眼底検査による眼科的評価は、適切に治療を行うために1-2年ごとに実施する。
- 空腹時血糖は継続的に検査し、テストステロン濃度(男性の場合)は、思春期が始まる前から、2-3年ごとに実施する。
- 適切な治療が行われていない成人など病気が進行している場合や、病気の後期においては、2-3年ごとに下記の検査を行う。
- 冠状動脈やその他の血管の石灰化を検出するための胸部CT検査
- 心電図検査(ECG)
- 大脳の萎縮や石灰化を評価するための頭部CT検査あるいはMRI
- 進行性筋力低下や嚥下困難を評価するための、筋電図検査(EMG)、口腔感覚運動検査、ビデオX線透視検査によるバリウム嚥下造影
- 肺機能検査
- 視覚-運動統合、視覚記憶、プランニング、注意の持続性、運動速度検査を含む神経および神経認知学的評価は、7-8歳から始める。
回避すべき薬物や環境
- 脱水は残存する腎機能をさらに悪化させる。
- 日光暴露は羞明を悪化させることがある。
リスクのある親族の検査
生命を脅かすシスチノーシスの合併症を防ぐために、早期に治療を開始する利点があるため、リスクのある親族(発端者の同胞の新生児など)のできるだけ早く検査をすることは、適切である。
その家系での遺伝子変異がわかっている場合は、分子遺伝学的検査、その家系での遺伝子変異がわかっていない場合は、生化学的検査を実施する。
リスクのある親族の検査に関連する問題については、"遺伝カウンセリングの目的"の項を参照。
妊娠経過管理
シスチノーシス患者の妊娠は、早産のリスク増加があり、周到にモニタリングしなければならない。腎移植後の女性では、腹腔内に移植された腎臓が、機械的な問題を生み出す。腎移植していない女性では、体液や電解質の状態の注意深い管理が必要である。
研究中の治療法
マウスにおいて、骨髄由来幹細胞移植が慢性腎不全を改善した。
他の幅広い疾患も含めた臨床研究の情報は"Clinical Trials.gov"を参照
その他
シスチノーシスの新生児マススクリーニング法の開発が、幅広い治療の成功を可能にすると期待されている。2つの方法が提案されている。:
- タンデムマススペクトロメトリーを用いた乾燥血液スポット中の七炭糖誘導体(セドヘプツロース)の同定。七炭糖(C7 sugar)誘導体は、シスチノーシスを引き起こす最も頻度の高い病因変異である57-kbホモ接合体欠損で検出される(訳者補足:57-kb欠失は、CTNSの近傍にあるセドヘプツロキナーゼをコードするSHPK (CSRKL)遺伝子を巻き込んでいるためその基質であるセドヘプツロースの尿中及び血中の濃度が高い)。
食物中の含硫アミノ酸制限、アスコルビン酸摂取およびジチオトレイトール(DTT)の使用は、効果がないことが証明されている。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
すべての型のシスチノーシスは、常染色体性劣性様式で遺伝する。
患者家族のリスク
発端者の両親
- 罹患者の親は、ヘテロ接合体であり、片方のアリルに変異を有する絶対保因者である(すなわち、CTNS遺伝子に1つの病因変異を有する保因者)
- ヘテロ接合体(保因者)は、臨床的に異常を認めない無症候性であり、病気を発症するリスクはない。
発端者の同胞
- 受精段階で罹患者の同胞が発症する確率は25%、発症せずに保因者となる確率は50%、発症もせず保因者ともならない確率は25%である。
- ヘテロ接合体(保因者)は、臨床的に異常を認めない無症候性であり、病気を発症するリスクはない。
発端者の子
- 罹患者の子供は、シスチノーシスを発症する病因変異を持った絶対保因者である。
- まれに、2世代で発症する(ときに、"偽優性"と呼ばれる)家族がいるが、これは罹患者が、CTNS遺伝子の病因変異を有するヘテロ接合体(保因者)パートナーとの間に子供ができた場合に生じる。
発端者の他の家族
-
発端者の親のそれぞれの同胞は、CTNS遺伝子の病因変異を有する保因者となるリスクは50%である。
保因者診断
生化学的検査:保因者検査は生化学的に実施することができる。その際には、新たに調整された白血球と適切なコントロールが必要である。
分子遺伝学的検査:リスクのある家族の保因者検査は、その家族内の病因変異がわかっている場合は可能である。
遺伝カウンセリングに関連した問題
早期診断、治療を目的としたリスクの高い親族に関する評価の情報については、"リスクの高い親族の評価"を参照
家族計画
- 遺伝的なリスクの識別,保因者であるか否か明確にすること、出生前診断が可能かについての討議は、妊娠前に時間をとって話しあうようにする。
- 若い世代の罹患者、保因者、保因者のリスクのある人に、(子への潜在的なリスクや生殖補助医療の選択などの議論も含めた)遺伝カウンセリングを提供することは適切である。
- シスチノーシスの女性が、妊娠し健康な新生児を出産したことがある。しかしながら、システアミンの胎児への催奇形性の影響について、人ではまだ研究がされていない。
- シスチノーシスの男性の妊孕性についてはデータがない。しかしながら、精巣のバイオプシーにより、精子は形成されているので、罹患者の精子凍結も考慮できるかもしれない。
DNAバンクは、将来の使用のために、(通常は白血球から抽出した)DNAを貯蔵しておくことである。検査手法や遺伝子、アリル変異、疾患への理解は将来改善する可能性があるため、罹患者のDNAを貯蔵しておくことは考慮されるべきである。
出生前/着床前診断
生化学検査
腎障害型シスチノーシスのリスクの高い妊娠に対する出生前診断は、絨毛膜絨毛あるいは羊水穿刺で採取した羊水細胞中の、いずれかのシスチン濃度を測定することのより可能である。
分子遺伝学的検査
罹患者の家族にCTNS遺伝子変異が同定されている場合は、シスチノーシスのリスクの高い妊娠に対する出生前診断と着床前診断は、両親が考慮してもよい選択肢となる。
訳注:日本では本症に対する出生前診断は行われていない。
専門医の間でも、家族の間でも、特に出生前診断を早期の診断というよりも妊娠中絶のための検査と考えているような場合は、考え方に相違があるかもしれない。多くの医療機関では、出生前診断について両親の意思を尊重することになるが、これらの問題に関して、議論されることが大切である。
表2.
GeneReviewで議論されたCTNS遺伝子の病因変異
| DNA Nucleotide Change | Predicted Protein Change | Reference Sequences |
|---|---|---|
| g.36,254_93,510del (57-kb del) 1 |
-- | AF168787 |
| c.18_21del4 | p.Thr7PhefsTer7 | NM_004937 .2 NP_004928 .2 |
| c.198_218del (198del21bp or 537del21)2 |
p.Ile67_Pro73del | |
| c.382C>T | p.Gln128Ter | |
| c.397A>T | p.Ile133Pro | |
| c.414G>A | p.Trp138Ter | |
| c.416C>T | p.Ser139Phe | |
| c.473T>C | p.Leu158Pro | |
| c.544T>C | p.Trp182Arg | |
| c.589G>A | p.Gly197Arg | |
| c.611_613del3 | p.Asp205del | |
| c.853-3C>G | --3 | |
| c.898_900+24del27 | -- | |
| c.922G>A | p.Gly308Arg |
注:表にある変異の分類は、著者らによって提供されたものであり、GeneReviewsのスタッフはその分類について、個別に確認していない。
注:命名法は、the Human Genome Variation Society (www .hgvs.org)の標準的な命名法に従っている。
- Touchman et al [2000]
- 現在の標準的な命名法には従っていない。
- Anikster et al [2000]
正常な遺伝子産物:CTNS遺伝子がコードするシスチノシンは、367個のアミノ酸からなり、7つの膜貫通部位、2つのリソソーム局在モチーフ、128個のアミノ酸からなる7つのN-結合型グリコシル化部位を有するN末端領域と、細胞質側に10個のアミノ酸からなるC末端領域を有する。シスチノシンはすべての組織の細胞に発現している。シスチノシンは、ジスルフィド結合アミノ酸であるシスチンを、リソソームから細胞質へ輸送する。シスチノシンは、マウスとヒトで高度に保存されている。
異常な遺伝子産物:CTNS遺伝子の57-kbの欠失を有すると、CTNS mRNAは作られない。一方、大部分の別のアリルは、mRNAの発現が残存している。重症型の患者においては、CTNS遺伝子に変異を有するアリルから、機能が欠失したシスチノシンが作られ、中間型あるいは眼型の患者においては、シスチノシンの機能が残存しているシスチノシンが作られていることが予想される。更新履歴
-
Gene Review著者: Galina Nesterova, MD and William A Gahl, MD, PhD.
日本語訳者: 岡村匡史、大熊喜彰、赤平百絵(国立国際医療研究センター)
Gene Review 最終更新日: 2014.1.30. 日本語訳最終更新日: 2016.9.20. -
Gene Reviews著者:Galina Nesterova, MD and William A Gahl, MD, PhD.
日本語訳者: 岡村匡史、大熊喜彰、赤平百絵(国立国際医療研究センター)
Gene Reviews 最終更新日: 2016.10.6.日本語訳最終更新日: 2017.2.3.(in present)
原文 Cystinosis