拡張型心筋症概説
(Dilated Cardiomyopathy Overview)
[Synonyms:OFD1,OrofaciodigitalSyndromeⅠ]
Gene Reviews著者: BRay E Hershberger, MD and Elizabeth Jordan, MS, LGC
日本語訳者:武井 眞(東京都済生会中央病院循環器内科)
GeneReviews最終更新日: 2022.4.7. 日本語訳最終更新日: 2023.7.12.
原文: Dilated Cardiomyopathy Overview
要約
本概説の目的は臨床家の拡張型心筋症(Dilated cardiomyopathy, DCM)の遺伝的背景に対する認識を高め、遺伝性DCMの症例に対して早期の診断と介入によるメリットを提供することである。
以下に本概説の目標を示す。
目標1.
DCMを定義すること
目標2.
DCMの分類を理解すること
目標3.
他の先天性症候群を伴わないDCM発端者の評価戦略を提供すること
目標4.
DCM発端者の無症状の親族に対して遺伝的リスクの評価を行い、心臓サーベイランスの提供、早期のHCMの診断および治療を可能とすることで長期予後を改善すること
1.拡張型心筋症の定義
拡張型心筋症は以下の二つの兆候の確認により診断される
- 左室拡大:左室拡大は通常成人において心臓超音波検査もしくは心臓MRI検査によって評価される。小児では発達の速度が速いため、小児患者において左室拡大を評価する際には専門家による評価を推奨する。
- 心筋収縮力の低下:左室駆出率(Left ventricular ejection fraction: LVEF)50%未満は収縮障害と考えられる。LVEFは臨床現場でもっとも一般的に用いられる収縮能の指標で、2次元心臓超音波検査もしくは心臓MRIにより評価される。非侵襲的な他の評価法として核医学検査がある。また、左室造影によってもLVEFを評価することができる。
注:主に左室に病変が認められる不整脈源性右室心筋症(Arrhythmogenic right ventricular cardiomyopathy: ARVC)はDCMと診断されることがある。[Sen-Chowdhry et al 2008]
DCMは通常40-60歳の成人で発症することが多いが、どの発生段階、年齢でも発症しうる(胎児期、新生児期、幼児期、小児期、老年期)。より詳細なDCMについての臨床的及び遺伝的情報は以下の文献を参照すること。[Burkett & Hershberger 2005, Sivasankaran et al 2005, Judge 2009, Dellefave & McNally 2010, Hershberger et al 2010a, Jordan & Hershberger 2021]
DCMの患者は年余にわたり無症状でありうる。臨床症状の発現は病期としては後期に起こることが多く、以下のような兆候を含む。
- 心不全:うっ血(浮腫、起坐呼吸、発作性夜間呼吸困難)や低心拍出(疲労、労作時の息切れ)などの症状
- 不整脈、刺激伝導系の異常:進行した心筋症、心不全では一般的に認められる。いくつかの原因遺伝子(DES, FLNC, LMNA,SCN5Aの病的バリアント)によるDCMでは左室機能低下の程度に比して不相応に高度の刺激伝導系の異常、不整脈を認めることがある。
- 血栓症:左室内血栓に続発する脳血管、もしくは全身への二次性塞栓症が起こりうる。
- 妊娠時の異常:妊娠中もしくは分娩直後に発症する周産期もしくは妊娠関連心筋症(Peripartum or pregnancy-associated cardiomyopathy : PPCM/PACM)はかつてDCMの中でも独立した疾患として認識されてきたが、近年ではDCMの臨床スペクトラムに連なるものとして認識されている。
2.拡張型心筋症の分類
DCMは後天性のDCM、DCMを表現型として含む先天性症候群(他臓器にも所見を有する)もしくは非症候群性(他臓器に所見を有さない)の先天性DCMに分類される(FIgure1)。
DCMは後天性のDCM、DCMを表現型として含む先天性症候群(他臓器にも所見を有する)もしくは非症候群性(他臓器に所見を有さない)の先天性DCMに分類される(FIgure1)。
後天性(二次性)DCM
後天性DCMのもっとも一般的な原因は冠動脈疾患からの心筋梗塞などの心筋虚血である。
その他のより頻度の低い原因として弁膜症、先天性心疾患、心毒性物質(アンスラサイクリンなどの抗がん剤、 この次訳まだ)、甲状腺疾患、炎症性疾患または感染症によるもの、長時間持続している高度の高血圧、放射線被ばくなどが挙げられる。近年抗がん剤暴露後に発症するDCMについても遺伝的背景が影響する可能性が示唆されているが、臨床的にはまだ議論のある部分である。[Garcia-Pavia et al 2019].
注:後天性DCMについては本概説ではこれ以上は扱わない。
先天性症候群に伴うDCM(他臓器にも所見を有する)
GeneReviewsでは、a)特定の診断名を示唆する(分子遺伝学的検査により確定されうる)もしくはb)確定的分子遺伝学的所見がない状態でも診断を確定できる一連の多臓器の臨床所見を示すものを症候群と定義する。先天性症候群に伴うDCMの一部のリストをTable1.に示す。[Hershberger et al 2009, Hershberger et al 2013].
注:本概説では先天性症候群に伴うHCMについて以下の表以上の言及は行わない。
表1. 先天性症候群に伴うDCM
| 疾患名 1 | 原因遺伝子 | 遺伝形式 | 臨床的特徴 | コメント |
|---|---|---|---|---|
| Barth症候群 | TAFAZZIN(TAZ) | XL |
|
|
| Carvajal 症候群 (OMIM 605676) |
DSP | AR | ||
| Duchene, Becker型筋ジストロフィ | DMD | XL | 男性において
|
ヘテロ接合の女性はDCMのみの表現型を呈することがある。 |
| Emery-Drefuss筋ジストロフィ | EMD FHL1 |
XL |
|
刺激伝導系の異常、不整脈のに頻度が高い |
| LMNA | AD/AR | |||
| HFEヘモクロマトーシス | HFE | AR |
|
非拡張型もしくは浸潤性心筋症の頻度がDCMよりも高い |
| Laing遠位型ミオパチー | NYH7 | AD |
|
|
| ミトコンドリア病に伴うDCM(ミトコンドリア病概説を参照) | mtDNA | Mat | 以下を含む複雑な表現型
|
AD = 常染色体顕性遺伝; AR = 常染色体潜性遺伝; Mat= 母系遺伝; XL = X連鎖
- アルファベット順
先天性DCM(先天性症候群に分類されない)
後天性DCMもしくは先天性症候群に伴うDCM(Table1)は先天性症候群に伴わない先天性DCM(GeneReviewでは他臓器病変を伴わないDCM)に含まれない。現在知られているDCM関連遺伝子について、ClinGen分類の強さ及びアルファベット順により並べたリストをTable2に示す。[Jordan et al 2021]
ClinGen DCM Gene Curation Expert PanelはClinGenフレームワークを用いて単一遺伝子による非症候群性DCMとの関連の強さを評価し、DCM関連遺伝子を分類している。それぞれの遺伝子についてまとめられたデータはClinGen Gne Validity Classificationから入手できる。
注:左室緻密化障害(Left ventricular non-compaction: LVNC)は左室心筋の形態的な特徴で、一般人でも認められるがDCMをはじめとしたほかの心血管疾患との関連も報告されている。しかしながら、LVNCとDCM表現型やその強さとの関連は以前不明である。 [Hershberger et al 2017, Ross & Semsarian 2018, Ross et al 2020]
表2.非症候群性DCM関連遺伝子
| 遺伝子1 | 遺伝形式 | DCMに占める当該病的バリアントの割合2 | ClinGenによる臨床的妥当性分類 | 特徴的な表現型 | 同一アレル疾患 3 | 参考文献/OMIM Gene Entry |
|---|---|---|---|---|---|---|
| TTN 4 | AD | 15-20% | Definitive | DCMはtruncating vatiantと関連する | 拡張型心筋症 早発性呼吸不全を伴う遺伝性ミオパチー 肢帯筋ジストロフィー2J Salih ミオパチー Udd 遠位型ミオパチー HCM |
188840 |
| LMNA | AD | 6% | Definitive | 不整脈、刺激伝導系異常 | 一部の例として Partial lipodystrophy シャルコーマリートゥース遺伝性ニューロパチー2B1 Emery-Drefuss筋ジストロフィ Hutchinson-Gilford早老症 |
150330 |
| FLNC | AD | 2-4% | Definitive | 不整脈、刺激伝導系異常 | Myofibrillarミオパチー 肥大型心筋症 拘束型心筋症 遠位型ミオパチー |
102565 |
| BAG3 | AD | 3% | Definitive | Myofibrillarミオパチー | 603883 | |
| TNNT2 | AD | 3% | Definitive | 肥大型心筋症 拘束型心筋症 |
191045 | |
| RBM205 | AD | 2% | Definitive | 不整脈、刺激伝導異常 DCMはエクソン9のホットスポットと関連 |
613171 | |
| SCN5A | AD | 2% | Definitive | 不整脈、刺激伝導系異常 | 一例として:QT延長症候群、Brugada症候群、特発性心室細動 | 600163 |
| DES | AD | <1% | Definitive | 不整脈、神経筋疾患 | 筋原線維性ミオパチー; Kaeser型神経性肩甲腓骨症候群 | 125660 |
| PLN | AD | <1% | Definitive4 | 不整脈、刺激伝導系異常 | 肥大型心筋症 | 172405 |
| TNNC1 | AD | <1% | Definitive | 肥大型心筋症 | 191040 | |
| DSP | AD | 不明 | Strong | 不整脈、刺激伝導系異常、右室への進展 | 不整脈源生右室心筋症、致死性アカントライティック表皮水疱症、掌蹠線状角化症(II)、皮膚脆弱-羊毛髪症候群、Carvajal症候群 | 125647 |
| ACTC1 | AD | <1% | Moderate | 肥大型心筋症 | 102540 | |
| ACTN2 | AD | <1% | Moderate | 肥大型心筋症 | 102573 | |
| TPM1 | AD | <1% | Moderate | 肥大型心筋症 | 191010 | |
| JPH26 | AD,AR | 不明 | Moderate | 肥大型心筋症 | 605267 | |
| NEXN | AD | 不明 | Moderate | 肥大型心筋症 | 613121 | |
| TNNI3 | AD, AR | <1% | Moderate | 肥大型心筋症 拘束型心筋症 |
191044 | |
| VCL | AD | 不明 | Moderate | 肥大型心筋症 | 193065 | |
| MYH6 | AD | 4% | Limited | 肥大型心筋症、心房中隔欠損症 | 160710 | |
| MYPN | AD | 3% | Limited | 拘束型心筋症、肥大型心筋症 ネマリンミオパチー |
608517 | |
| MYBPC3 | AD | 2% | Limited | 肥大型心筋症 | 600958 | |
| CSRP3 | AD | <1% | Limited | 肥大型心筋症 | 600824 | |
| ILK | AD | <1% | Limited | 602366 | ||
| LAMA4 | AD | <1% | Limited | 600133 | ||
| LDB3 | AD | <1% | Limited | 肥大型心筋症、筋原線維製ミオパチー | 605906 PS601419 |
|
| PSEN2 | AD | <1% | Limited | 早発性アルツハイマー認知症 | 600759 | |
| SGCD | AD | <1% | Limited | 肢帯型筋ジストロフィ2F/R6 | 601411 | |
| TCAP | AD | <1% | Limited | 肥大型心筋症 肢帯筋ジストロフィ2G/R7 |
604488 | |
| ABCC9 | AD | 不明 | Limited | 家族性心房細動、Cantu症候群(多毛症を伴う骨軟骨異形成症) | 601439 | |
| ANKRD1 | AD | 不明 | Limited | 609599 | ||
| CTF1 | AD | 不明 | Limited | 600435 | ||
| DSG2 | AD | 不明 | Limited | 右室進展の可能性 | 不整脈源生右室心筋症 | 125671 |
| DTNA | AD | 不明 | Limited | 先天性心奇形 | 601239 | |
| EYA4 | AD | 不明 | Limited | 難聴 | DFNA10非症候群性難聴 | 603550 |
| GATAD1 | AR | 不明 | Limited | 614518 | ||
| MYL2 | AD | 不明 | Limited | 肥大型心筋症 | 160781 | |
| NEBL | AD | 不明 | Limited | 605491 | ||
| NKX2.5 | AD | 不明 | Limited | 先天性心奇形 | 600584 | |
| OBSCN | AD | 不明 | Limited | 608616 | ||
| PLEKHM2 | AR | 不明 | Limited | 609613 | ||
| PRDM16 | AD | 不明 | Limited | 605557 | ||
| TBX20 | AD | 不明 | Limited | 心房中隔欠損 | 606061 |
|
| TNNI3K | AD | 不明 | Limited | 不整脈、刺激伝導系異常 | 刺激伝導系異常 | 613932 |
AD = 常染色体顕性遺伝; AR = 常染色体潜性遺伝
- 表の順序は臨床的妥当性分類、DCMの原因としての頻度、アルファベット順に従った。
- 呈示された頻度(DCM発端者を多数スクリーニングした2件以上の報告に基づく)は、予備的なものとして解釈すること。
- 同一遺伝子の病的バリアントによって引き起こされる他の表現型
- 注:
DCMの3つのコホートの報告(家族性、孤発例いずれも含む)によると、10-20%のDCMはTTNの病的なトランケーションバリアントを持つとされる[Herman et al 2012]が、TTN病的バリアントのDCMにおける役割を決定するのは以下の理由から困難である。a)健常コントロールの3%がトランケーションバリアントを有する、b)TTNの病的トランケーションバリアントはすべての家系でDCMの発症と一致した分離を示すわけではない[Norton et al 2013]。DCM罹患者から同定されたTTNのトランケーションバリアントはタンパクコーディング領域であるA-band領域に集積していることも報告されている[Roberts et al 2015]。今日まで、DCMとの関連が報告されているTTNのミスセンスバリアントはない。 - RBM20の病的もしくは病的である可能性が高いDCM関連バリアントはエクソン9に集積している。エクソン9以外のRBM20のバリアントがDCMと関連するかどうかは不明である。
- Sabater-Molina et al [2016], Miura et al [2020]
3.(可能な場合における)拡張型心筋症の特定の遺伝的背景の確立
遺伝学的検査はPPCM/PACMを含む非虚血性DCMのすべての症例について年齢にかかわらず提供されるべきである。[Goli et al 2021](Figure1)現状のDCM関連遺伝子についてはTable2を参照すること。DCMの遺伝学的診断を確立する目的は発端者の血縁者についてリスク評価を提供するためである。(4章を参照)
家族性DCM症例(家系内の2親等以内にDCM症例)や孤発例DCMの30-35%程度までに30以上の遺伝子のバリアント(病的、病的である可能性が高いもしくはVUS)が報告されている。[Hershberger et al 2013, Jordan & Hershberger 2021] [Hershberger et al 2008, Hershberger et al 2010b, Pugh et al 2014, Morales et al 2020]病的もしくは病的である可能性が高いバリアントの検出率は27%程度とされている。[Pugh et al 2014]
ClinGen分類でdefinitive, strong, moderate 以上(Table2参照)の遺伝子を含む心筋症マルチ遺伝子パネル検査が遺伝的原因の同定できる確率が高く、同時に意義不明のバリアント(VUS)や病態を説明しえない病的バリアントが同定される頻度を制限することが可能である。
注:1)検査機関によってパネルに含まれる遺伝子の種類、検査精度が異なる可能性がある。また、これらの事柄は経時的にも変化すると考えられる。2)いくつかのマルチジーンパネルにはGeneReviewで取り上げている病態と関連しない遺伝子が含まれている可能性がある。3)いくつかの検査機関ではパネルオプションとして検査機関によってデザインされたパネルや臨床家によって指定された遺伝子を含む表現型に注目したエクソーム解析を提供している。4)パネル検査で行われる解析手法にはシークエンス、欠失/重複解析、シークエンスを用いない遺伝子解析が含まれる。
マルチジーンパネルについての基本的な情報はこちらを、遺伝学的検査を依頼する臨床家に向けた詳しい情報はこちらを参照。
遺伝学的検査を依頼する医療従事者はDCMの遺伝学に精通していることが求められる。
[Burkett & Hershberger 2005, Judge 2009, Caleshu et al 2010, Hershberger et al 2010a, Hershberger & Siegfried 2011, Hershberger et al 2013, Jordan et al 2021].遺伝学的検査の結果解釈は複雑で、患者のサーベイランスや臨床的マネジメントに及ぼす影響を考慮すると心血管遺伝センターや心血管疾患の遺伝学に精通した遺伝カウンセラーへの紹介を検討する必要がある。(NSGC-Find a Genetic CounselorやABGC Find a Certified Genetic Counselorなどで検索可能)
4.リスク親族において早期治療可能なDCMの表現型を検知するための遺伝的リスク評価
DCM患者の無症候の一親等の血縁者における心血管スクリーニングは早期のDCMの検知、迅速な治療の開始を可能とし、長期予後を改善するとされる。[Morales & Hershberger 2015]. DCM患者の一親等以内の血縁者において遺伝学的背景を明らかにすることでリスクのある親族がだれなのか、それ以降の心血管スクリーニングの頻度について情報を得ることができる。[Hershberger et al 2018].
非症候群性のDCMにおけるリスク親族に対する遺伝的リスクの評価、心血管サーベイランスについての基本的な考え方を本章で提示する。それぞれの家系、遺伝子に特異的な事象については完全に網羅することは本章では行っていない。
注:
発端者がDCMを表現型として含む先天性の症候群(Barth症候群、Duchene型筋ジストロフィなど)を呈している場合、その症候群に対するカウンセリングを施行すること(Table1を参照)。こうした先天性の症候群に伴うDCMについては本概説ではこれ以上扱わない。
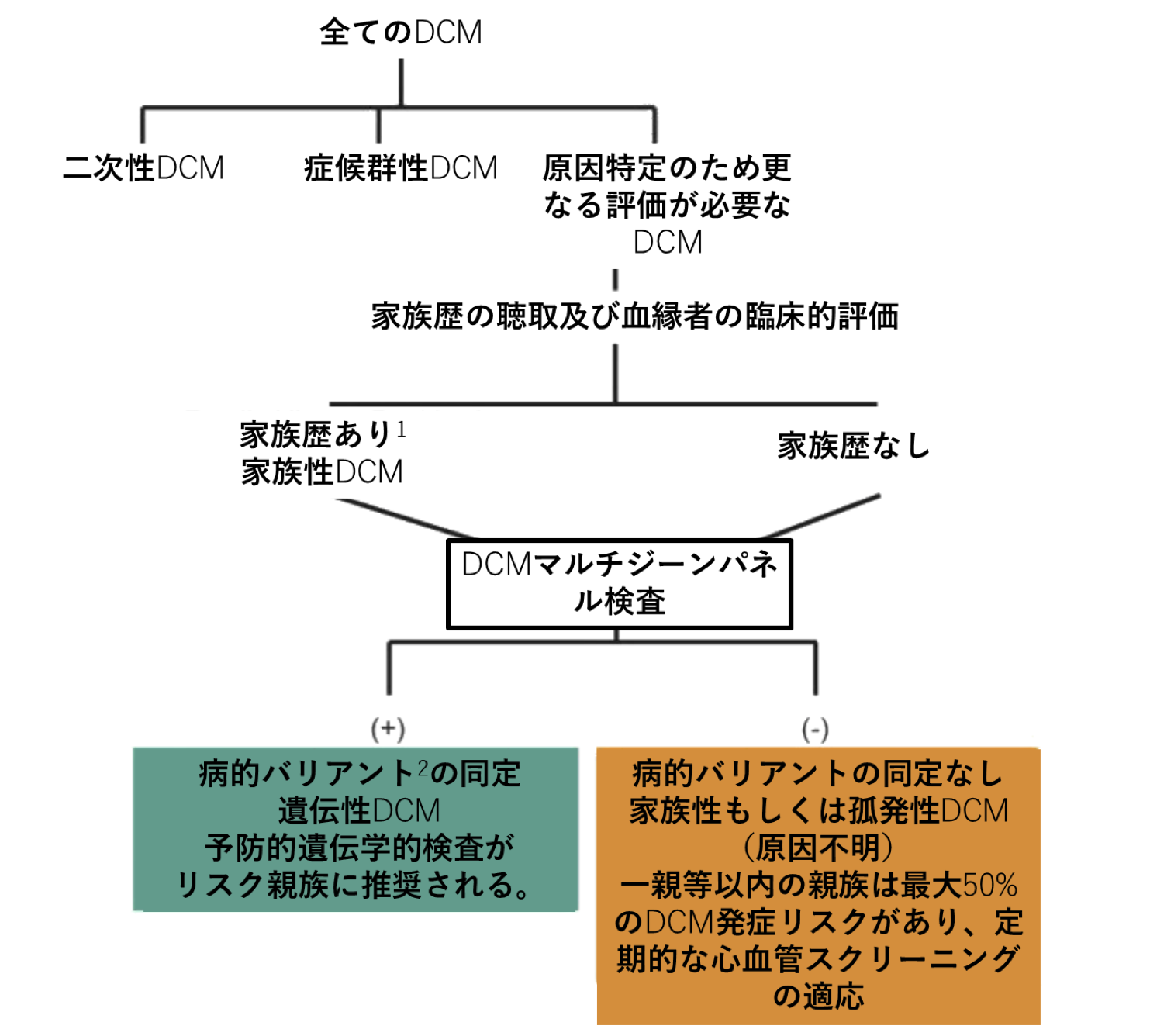
図1:DCMの分類
注:
- 発端者を含み家系内に二人以上のDCM患者が同定されること
- ACMG/AMP分類に従う
遺伝リスクの評価
遺伝カウンセリングとは、個人及び家族に対して、遺伝性疾患の性質、遺伝の 様態及び遺伝の意味について情報を提供し、十分な情報に基づいた医療上及び個 人的な意思決定を支援するプロセスである。以下のセクションは、遺伝的リスク評価、家族の遺伝的状態を明らかにするための家族歴と遺伝学的検査の利用を扱っている。-ED。
非症候群性DCMは、一般的に常染色体優性遺伝する。JPH2およびTNNI3関連非症候群性DCMは常染色体優性遺伝または常染色体劣性遺伝をする。
DMD関連DCMは、男性では症候群性DCMとして、保因者の女性では孤立性DCMとして現れることがあり(Table1)、X連鎖的に遺伝する(ジストロフィノパチーの項を参照)。
発端者の同胞
同胞のリスクは発端者の両親の臨床的、遺伝的状態による.
- 発端者のどちらかの親が病的もしく病的である可能性が高いバリアントを有していた場合、同胞が病的アレルを継承するリスクは50%である.表現型が多様であること、浸透率が低いことから臨床的重症度と発症年齢を予測することは困難である。
- もし発端者の両親がDCM関連の病的もしくは病的である可能性が高いバリアントを有している場合、同胞が一つ以上のDCM関連バリアントを継承する可能性は75%で、病的バリアントを一つも継承しない可能性は25%である。
- 仮に両親が臨床的に無症状であっても、両親の遺伝的背景が不明の場合、浸透率の低さに加え、性腺モザイクの可能性もある
常染色体潜性遺伝
発端者の両親
- ある罹患者の両親は定義上ヘテロ接合(JPH2もしくはTNNI3の病的バリアント保因者と想定される)である。
- 両親ともに JPH2 または TNNI3 病原変異体のヘテロ接合体であることを確認し、信頼性の高い再発リスク評価を可能にするために、発端者の両親に対して分子遺伝学的検査を行うことが推奨される。片方の親にのみ病原性変異体が検出され、生物学的母子関係および父子関係が確認された場合、以下の可能性を考慮すべきである:
- 発端者で同定された病的バリアントのひとつが、発端者の代でde novoに発生したか、モザイクの親で接合後de novoイベントとして発生した [Jónsson et al 2017]。
- 病的バリアントを有する親染色体の片親性イソダイソミーにより、発端者において病的バリアントのホモ接合が生じた。
JPH2またはTNNI3の両アレル変異を持つ子どものヘテロ接合体の両親はDCMのリスクがある可能性がある。JPH2関連DCMおよびTNNI3関連DCMは常染色体潜性遺伝または常染色体顕性遺伝する可能性があり、それぞれの病的バリアントがどちらの遺伝形式をとっているか区別することは現状できない。従って、発症した患者の両親が、発症した患者に同定された2つの病的バリアントのうち1つしか持っていない場合でも、心臓サーベイランスを受けることは妥当である。
発端者の同胞
- 両親ともにJPH2またはTNNI3のヘテロ接合体であることが分かっている場合、罹患者の兄弟姉妹は受胎時に25%の確率で両アレルに病的バリアントを受け継ぎ、50%の確率で発端者に同定される2つの病的バリアントのうち1つを受け継ぐ。どちらの病的バリアントも受け継がない可能性は25%である。
- 発端者で同定された病的バリアントの1つをヘテロ接合体で持つ同胞は、DCMのリスクがある可能性がある。JPH2関連DCMおよびTNNI3関連DCMは常染色体潜性遺伝または常染色体顕性遺伝する可能性があり、それぞれの病的バリアントがどちらの遺伝形式をとっているか区別することは現状できない。従って、発症した患者の両親が、発症した患者に同定された2つの病的バリアントのうち1つしか持っていない場合でも、心臓サーベイランスを受けることは妥当である。
発端者の子供
常染色体劣性JPH2またはTNNI3関連DCM患者の子供は、定義上JPH2またはTNNI3の病的バリアントのヘテロ接合体であり、DCMのリスクがある可能性がある。したがって、子供はDCMの心臓サーベイランスを受けるべきである。
心血管系サーベイランス
DCM発症リスクを有する家系内の個人に対して疾患の発症前に臨床的、遺伝的評価を行うことは、無症候性のDCM診断、未発症者の早期診断を行うことでを疾患晩期の症候性病期への進展を予防、もしくは遅延させることが可能となり、適切である。[Morales & Hershberger 2015].
発端者がDCM関連遺伝子に既知の病的バリアントを有する場合
Figure1の青緑の枠を参照すること(病的バリアントが同定されている場合)。発端者の両親、同胞、子供、リスク親族について遺伝的診断を明らかにするために分子遺伝学的検査が推奨される。
家族性DCM関連病的バリアントが同定された血縁者については生涯でのDCM発症リスクが高くなる。無症候性である場合、個々の年齢に併せて臨床的な心血管スクリーニングを受けるべきである。[Hershberger et al 2018].
注:
無症候のリスク親族では、DCMの臨床診断基準を満たさない症例であっても、心臓超音波検査の解釈があいまいである場合(左室拡大はあるが左室収縮は良好である場合、逆に左室拡大はないが左室収縮能が低下している場合等)や心臓超音波検査が正常であっても心電図に異常がある場合(伝導障害や不整脈)が認められる場合、早期のDCMである可能性がある。
一般的に、発端者に同定された病的バリアントが検出されない血縁者についてはDCMの障害発症リスクは上昇していないと考えられるため、心血管スクリーニングの必要はない。しかしながら、非症候群性DCMの患者にはDCM関連遺伝子に複数のバリアントが検出されることがあり、血縁者ではこうした複数のバリアントが分離して検出されることがあるため、臨床所見、遺伝的背景、家族歴を考慮した個別のリスク評価を行い、心血管サーベイランスが不要かどうかを判断する必要がある。[Morales et al 2020] [Liu et al 2015, Cowan et al 2018].
発端者においてDCMの特異的な遺伝的原因が特定されていない場合
Figre1のオレンジの枠(病的バリアントが同定されない場合)を参照すること。無症候のリスク親族に対して年齢に応じた間隔で心血管スクリーニングを行う。
注:
無症候のリスク親族では、DCMの臨床診断基準を満たさない症例であっても、心臓超音波検査の解釈があいまいである場合(左室拡大はあるが左室収縮は良好である場合、逆に左室拡大はないが左室収縮能が低下している場合等)や心臓超音波検査が正常であっても心電図に異常がある場合(伝導障害や不整脈)が認められる場合、早期のDCMである可能性がある。
発端者の一親等以内のリスク親族がDCMと診断された場合、家族性DCMの診断となり、サーベイランスの範囲を新たにDCMと診断された親族の一親等以内の親族にも拡大することが推奨される。
以下の場合は将来再度の遺伝学的検査が発端者(及び遺伝的背景を把握することが有益な情報を提供する可能性のある親族)に行うことを考慮する。
- マルチジーンパネル検査に搭載される遺伝子の数が増える、もしくは感度が上昇する場合
- ゲノムワイド検査(エクソームもしくはゲノムシークエンシング)が臨床的に使用可能となった場合
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- American Heart AssociationDilated Cardiomyopathy (DCM)
Cardiomyopathy UK
United Kingdom
Phone: 0800 018 1024 (UK only)
Email: contact@cardiomyopathy.org
www.cardiomyopathy.org
- Children's Cardiomyopathy Foundation Phone: 866-808-2873 (toll-free)
- DCM Foundation Phone: 833-DCM-HOPE (833-326-4673)
- MedlinePlus Familial dilated cardiomyopathy
- Dilated Cardiomyopathy Research Project hone: 877-800-8430
Fax: 201-227-7016
Email: info@childrenscardiomyopathy.org
www.childrenscardiomyopathy.org
Email: Info@DCMFoundation.org
www.dcmfoundation.org
Email: DCM.Research@osumc.edu
www.dcmproject.com
更新履歴:
- GeneReviews著者:Ray E Hershberger, MD, Jessica D Kushner, MS, CGC, Sharie B Parks, PhD
日本語訳者:窪田美穂(ボランティア翻訳者),櫻井晃洋(信州大学医学部附属病院遺伝子診療部)
GeneReviews 最終更新日: 2008.7.10. 日本語訳最終更新日: 2009.3.19. - Gene Review著者: Ray E Hershberger, MD,Ana Morales, MS, CGC
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: : 2013.5.9. 日本語訳最終更新日: 2013.9.25 - Gene Reviews著者: Ray E Hershberger, MD and Ana Morales, MS, LGC.
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Reviews 最終更新日: 2015.9.24 語訳最終更新日: 2017.11.27 ( -
Gene Reviews著者: BRay E Hershberger, MD and Elizabeth Jordan, MS, LGC
日本語訳者:武井 眞(東京都済生会中央病院循環器内科)
GeneReviews最終更新日: 2022.4.7. 日本語訳最終更新日: 2023.7.12.[in present]
原文: Dilated Cardiomyopathy Overview
![]()