古典型エーラス-ダンロス症候群
(Classic Ehlers-Danlos Syndrome)[類義語:エーラス-ダンロス症候群,古典型EDS,古典型]
Gene Reviews著者: TFransiska Malfait, MD, PhD, Richard Wenstrup, MD, and Anne De Paepe, MD, PhD
日本語訳者: 與那嶺 関屋智子(金沢大学遺伝カウンセリングコース) ,渡邉淳(金沢大学附属病院遺伝診療部)
Gene Reviews 最終更新日: 2018.6.26 日本語訳最終更新日: 2020.7.19
原文: Classic Ehlers-Danlos Syndrome
要約
疾患の特徴
古典型エーラス-ダンロス症候群(古典型EDS)は皮膚過伸展,萎縮性瘢痕,ならびに全身関節可動亢進を特徴とする結合組織疾患である.皮膚は平滑,ビロード状の感触があり,過伸展すなわち簡単につかむことができよく伸びるが離すと直ちに元に戻る(皮膚弛緩症で見られる弛緩してたゆんだ皮膚とは異なる).皮膚は脆弱であり,特に圧がかかりやすい部位(膝, 肘)やけがをしやすい部位(脛, 額, おとがい)では軽度の外傷で裂けやすい.創傷治癒は遅れ,一次創傷治癒後の瘢痕形成が特徴的である.関節可動亢進によっておきる肩関節,膝蓋,指関節,股関節,橈骨および鎖骨の脱臼は,多くの場合自然に修復するか,あるいは患者自身で容易に対応できる.その他の特徴として運動発達遅延を伴う筋緊張低下,易疲労性,筋攣縮,易出血性が見られることがある.僧帽弁逸脱はまれに見られるが,臨床的にほとんど影響を及ぼさない. 大動脈起始部の拡張が報告されているが,若年に多く出現し,進行は稀である.
診断・検査
古典型EDSの診断は,臨床診断基準(皮膚過伸展,萎縮性瘢痕に加えて,全身関節可動亢進または,少なくとも3つの小基準症状)を最小限に認め,かつ遺伝学的検査でCOL5A1,COL5A2,(まれにCOL1A1)の病的変異をヘテロ接合体に同定された場合に確定診断される.
臨床的マネジメント
症状の治療:
古典型EDSの小児で筋緊張低下や運動発達遅延を来している場合,理学療法プログラムは有用である.体重負荷のない運動は筋肉の発達や協調を促す.抗炎症薬は関節痛を軽減することがある.筋緊張低下や慢性的な疼痛を伴う関節不安定性のある方は,症状に合わせた生活様式への適応が必要かもしれない.皮膚創傷は張力をかけずに通常2層に縫合する.他の外傷に対しては,深層部の縫合を行う.すなわち皮膚に隣接している領域を通常の2倍にする.隣接する皮膚との境界は瘢痕の伸展を防ぐために丁寧にテープを貼る.出血時間の正常化にはDDAVPⓇ(酢酸デスモプレシン)が有用であると考えられている.心血管系症状は標準的な治療を行う.
一次病変の予防 :
皮膚脆弱性がある年少児では,皮膚裂傷を裂けるため額,膝,脛には防護用パッドや包帯を着用すると良い.年長児は,活動する時にむこうずねのパッドが付いているサッカー用のパッドあるいはスキー用のストッキングを着用するとよい.アスコルビン酸 (ビタミンC) は出血傾向を軽減することがある.
経過観察大動脈拡張や僧帽弁逸脱を認めた場合は,年に1度の定期的な心エコー検査.
回避すべき薬物や環境:
関節を伸展させるスポーツ,アセチルサリチル酸(アスピリン)
遺伝カウンセリング
常染色体優性遺伝形式.患者の約50%は罹患した親から病的変異を受け継ぎ,罹患者の約50%は新生突然変異である.古典型EDSの子どもが病的変異を受け継ぐ割合は50%である. 病的変異が見いだされている罹患家系においては,ハイリスク妊娠のための出生前診断や着床前診断が可能である.
訳注:一般に古典型 EDSに対して出生前診断の適応があるとは考えられておらず,日本では行われていない.
診断
臨床診断
古典型EDSの診断は臨床検査と家族歴によってなされる.診断基準はVillefrancheで開催された会議で医学諮問グループによって決定された[Beighton et al 1998].そして最近国際EDSコンソーシアム[Malfait et al 2017]によって改訂された.
- 大基準は診断上の特異性が高い.大多数の罹患者にみられる症状であり,かつ/または本疾患に特徴的であり,他のEDS病型および/または他の遺伝性結合組織疾患との鑑別が可能となる.
- 小基準の症状は,古典型EDSを疑う重要な所見であるが,確定診断には不十分である.
古典型EDSの診断における大基準
- 皮膚過伸展 (図1)
- Remvigら[2009]に記載されているように,非利き腕の前腕の中央部の掌側表面の皮膚および皮下層をつまんで持ち上げることによって測定する.
- 皮膚は以下の3部位でのカットオフ値(前腕の遠位部と手の甲は1.5cm,首,肘,膝は3cm)よりも伸びると過伸展とされる.
- ,萎縮性瘢痕 (図2)
- 全身関節可動亢進 関節可動亢進(図3)は年齢,性別,家族歴および民族的背景によって異なる.
- 母指の過屈曲による前腕との接触:関節可動亢進の特徴
- 古典型EDSにみられる関節可動亢進は,全身的で 大関節,小関節両方にみられる.小児が歩き始める際に気づかれることが多い.
- 関節可動亢進を客観的に半定量的に測定するには,最も受け入れられている方法であるBeighton判定基準で評価する.(表1 参照).
- 関節の緩みは加齢と共に減弱するのでBeighton判定基準でスコアが5未満の人は,過去の観察に基づ いて陽性とみなされることがある. (5点法を参照)
| 関節/所見 | 陰性 | 片側 | 両側 |
|---|---|---|---|
| 小指の90度を超える過背屈 | 0 | 1 | 2 |
| 母指の過屈曲による前腕との接触 | 0 | 1 | 2 |
| 肘関節の10度以上の過伸展 | 0 | 1 | 2 |
| 膝関節の10度以上の過伸展 | 0 | 1 | 2 |
| 膝伸展位で脊柱を前屈させ手掌が床につく | 0 | 1 | |
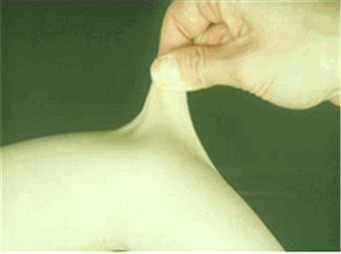 |
図1 |
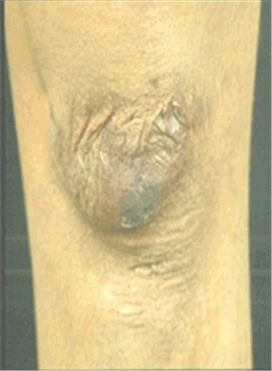 |
図2 |
 |
図3 |
5点以上で全身関節可動亢進とみなされる.
5点法(Hakim & Grahame [2003])
- 現在(または過去に),膝伸展位で手掌を床につくことができますか(できましたか)?
- 現在(または過去に)母指を曲げて前腕に触れることができますか(できましたか)?
- 子供の頃,体を変な形に歪ませたり開脚座(注釈:尻を床につけ両足を一直線に広げる)ができましたか?
- 子供,もしくは10代の頃,肩や膝が脱臼したことが複数回ありましたか?
- あなたは自分を「二重関節」(注釈:関節可動亢進) があると思いますか?
注:「はい」が2項目以上の場合,感度80-85%,特異度80-90%で関節可動亢進が示唆される.
古典型EDSの診断における小基準
- 易出血性
- 平滑でビロード状感触の皮膚
- 皮膚脆弱性(あるいは外傷性の裂傷)
- モルスクム様偽腫瘍 : 肘や膝のような圧迫される瘢痕部位で認める塊状の病変
- 皮下球状塊(subcutaneous spheroids):前腕やむこうずねの皮下組織の骨の突起部全体でみられる小さく,固い小結節状で可動性がある球状塊.レントゲン像では外側層は石灰化が見られ,中心部は透化している.
- ヘルニア(その既往歴)
- 内眼角贅皮
- 関節可動亢進の合併症 (すなわち,捻挫,脱臼/亜脱臼 疼痛,扁平足)
- 診断基準を満たす第一度近親の家族歴
- 大基準 皮膚過伸展と萎縮性瘢痕がみられる
- 大基準 全身関節可動亢進 および/または 3つ以上の小基準がみられる
診断の確立
古典型EDSの診断は,臨床診断基準の最小限を満たし,表2に列挙した遺伝子の1つに病的変異をヘテロ接合体で有することが確認された場合に確定診断される. 遺伝学的検査のアプローチには,単一遺伝子の同時(または段階的)検査,多遺伝子パネル,及びより網羅的なゲノム検査などがある.
- 同時単一遺伝子検査
- COL5A1,COL5A2,COL1A1を含む関連遺伝子群多遺伝子パネル検査(鑑別診断参照)
注(1)パネルに含まれる遺伝子や,各遺伝子に用いられる検査の診断感度は検査室によって異なり,時間を経ると変化する可能性がある.(2) 多遺伝子パネル検査の中には,本GeneReviewで検討されている病態とは関連のない遺伝子を含むものもあるので,臨床医は意義不明変異や,表現型を説明できない遺伝子の同定を少なくするため,最も合理的なコストで病態の遺伝的原因を同定する可能性が高い遺伝子パネルを決める必要がある.
(3) 検査室によっては,パネルのオプションとして,臨床医によって指定された遺伝子を含むパネル及び/または表現型に焦点をあてたエキソーム解析を含むことができる.(4)パネルで使用される方法は,塩基配列解析,欠失/重複解析,および/または塩基配列ではない検査を含む.
多遺伝子パネルはこちらを参照.
遺伝学的検査を検討される臨床医はこちらを参照.
- エキソーム解析および全ゲノム解析を含むより網羅的遺伝子検査(可能な場合)が検討されることがある. このような検査は,これまで想定されていなかった診断をもたらしたり,示唆されたりすることがある. (例えば,類似の臨床症状を示す異なる遺伝子(群)の病的変異) 網羅的遺伝子(ゲノム)検査はこちらを参照. 遺伝学的検査を検討される臨床医はこちらを参照.
注:病的変異が見つからない場合,COL5A1nullアレル検査でⅤ型コラーゲンの異常が示されることがある.検査を受けられるのは,COL5A1の多型マーカーにおいてヘテロ接合体を有した方に限定される. 皮膚生検からcDNAを解析し,片方または両方のマーカーの存在を調べる.片方のマーカーが検出できない場合,そのアレルは機能していない(すなわちnull)であると考えられる.この種の検査は広くは行われていない.
| 遺伝子1,2 | この遺伝子の病的変異に起因するcEDSの割合 | 手法による病的変異の検出可能な割合3 | |
|---|---|---|---|
| 塩基配列解析4 | 遺伝子欠失・重複解析5 | ||
| COL1A1 | <1%6 | 100%6 | 報告なし6 |
| COL5A1 | 75-78%7 | 99%7 | 1%7 |
| COL5A2 | 14%7 | 100%7 | 不明8 |
| 不明 | <10%7 | ||
- 遺伝子はアルファベット順に記載
- 表A(遺伝子ならびに染色体遺伝子座とタンパク名のデータベース)参照
- アレル変異の情報に関する「遺伝学的検査」の項を参照
- 塩基配列解析の結果は「良性」「おそらく良性」「意義不明」「おそらく病原性」「病原性」と解釈される
- 遺伝子欠失/重複解析は,遺伝子内欠失または重複を検出する.塩基配列解析では検出できないエクソンや全ゲノムの欠失を検査する,定量PCR法,ロングPCR法,MLPA法,標的マイクロアレイ法など,様々な方法がある
- Brady et al [2017], Malfait et al [2017
- Symoens et al [2012], Ritelli et al [2013]
- 8.遺伝子欠失/重複解析の検出率に関するデータはない.
注:遺伝学的検査ができない場合,皮膚生検での透過型電子顕微鏡(TEM)像でのコラーゲンフラワー所見(注釈:膠原線維の横断面で,異常な形をした大型線維が,正常線維間に混ざってみられる所見)は,確定ではないが診断の一助となる.
臨床像
自然経過
cEDSは,皮膚過伸展,創傷治癒の異常および全身関節可動亢進を特徴とする結合組織疾患である.これまで重症度のみでI型EDSとII型EDSの2つの亜型に分けられていたが,臨床所見は連続性があり現在1つの臨床病型として認識されている.
皮膚
皮膚過伸展は,(特に古典型 EDSでは)EDSの主症状の1つである.容易によく伸びるが離すと直ちに元に戻る(皮膚弛緩症における弛緩してたゆんだ皮膚とは異なる)
皮膚は平滑で ビロード状の感触がある.
皮膚は脆弱であり軽度の外傷で裂けやすく,特に圧がかかりやすい部位(膝, 肘)やけがをしやすい部位(脛, 額, おとがい)に多い. 皮膚脆弱性のため,皮膚や粘膜の縫合創の離開を起こしうる.
創傷治癒は遅れ,一見したところ正常な一次創傷治癒の後,瘢痕形成するのが特徴的である.瘢痕は大きくシガレットペーパー様(あるいは紙状)の形態になる.
- 古典型 EDSにみられる他の皮膚症状
- モルスクム様偽腫瘍(Molluscoid pseudotumors)
- 皮下球状塊(Subcutaneous spheroids)
- 圧迫性丘疹(Piezogenic papules):起立時に踵の正中側方に筋膜から真皮に向けた脂肪織がヘルニアを起こし,小さいが痛みを伴う.
- 蛇行性穿孔性弾性線維症(Elastosis perforans serpiginosa): 紅斑性の丘疹が蛇行状または環状に拡大するとともに,中心部は萎縮した特徴をもつ,原因不明のまれな皮膚状態.
- 肢(先)端チアノーゼ(Acrocyanosis): 皮膚(主に手)の微小血管の圧迫や狭窄によっておこる無痛性病変で,患部は青くなり,冷たく汗ばみ,局所的に腫脹を来す.
- 凍瘡(しもやけ) (Chillblain): 赤く腫脹する皮膚を特徴とする寒冷障害.皮膚を寒冷にさらして2時間以内に進行し,熱く掻痒を伴う.
組織脆弱性
広範囲の組織過伸展性,脆弱性による徴候は多臓器においてみられている.
- 妊娠中の子宮頚管不全
- 鼡径ヘルニアや臍ヘルニア
- 裂孔ヘルニアや腹壁瘢痕ヘルニア
- 小児期早期からの繰り返す肛門脱
関節
関節可動亢進による合併症.肩関節,膝蓋骨,指骨,股関節,橈骨や鎖骨の脱臼を生じ,多くは自然に修復し患者自身で簡単に対応できる.罹患者は骨レントゲン像で問題はなくても慢性の関節痛や下肢痛で悩まされることがある.
関節可動亢進に関連した他の問題として,関節不安定,先天性の内転内足,偏平足,側頭下顎関節の機能不全,関節滲出液および骨関節炎が挙げられる. [Hagberg et al 2004, De Coster et al 2005a, De Coster et al 2005b].
神経学的所見
原発性の筋緊張低下が起こり,運動発達遅延,歩行障害や軽度の運動障害を生ずることがある.疲労や筋攣縮は比較的頻繁に起こる.まれに,起立性低血圧や頭痛を引き起こす脳脊髄液の漏出が報告されている. [Schievink et al 2004].
易出血性
易出血性は頻繁にみられ,知らないうちに出血斑(自然出血)をとして現れる.特に脛や膝などの露出した部位では,頻繁に同一部位で生じ,皮膚は褐色変化が認められる.正常な凝固状態にもかかわらず,歯磨き時など出血が止まりにくい傾向がある.
心血管系
古典型EDSでは,構造的心奇形はまれである.
僧帽弁逸脱や(頻度は少ないが)三尖弁逸脱が起こることがある.僧帽弁逸脱の診断は厳格な診断基準に則る.
僧帽弁逸脱は,臨床的にほとんど影響がない傾向がある.[Atzinger et al 2011]
大動脈起始部拡張は古典型EDSで報告されている[Wenstrup et al 2002, McDonnell et al 2006, Atzinger et al 2011].
若年層に多くみられ,進行することはほとんどない.[Atzinger et al 2011]
古典型 EDSの重症型では,まれに頭蓋内動脈瘤や動静脈瘻に加えて大動脈の自然破裂が起こることがある.
妊娠
古典型EDS女性の妊娠では,妊婦のみならず新生児にも注意が必要である(参照 妊娠期の管理).全体的に見て古典型EDS女性の妊娠における合併症は通常妊娠に比べて多いが,これまでに良い調査研究はなく,各合併症の発生率の評価が難しいのが現状である.
- 母体が罹患している場合,早期破水や早産となることが多く,特に胎児が罹患している場合,重症度が最も高くなる.
- 児が罹患している場合,筋緊張低下のため骨盤位の頻度が高く,新生児期の股関節や肩関節の脱臼を起こすことがある.
数は限られているが,COL5A2の病的変異は,古典型EDSの臨床症状がより重症になると考えられている.
浸透率
浸透率は100%,あるいはそれより低いかは不明である.男女差はないと推定される.
EDS古典型の新しい命名は古典型EDS,またはcEDSである.[Malfait et al 2017].
頻度
古典型EDSの頻度は1:20 000と推定されている.従来II型EDSに分類されていた軽症者は,医療的問題にならないこともあり,気づかれない.
まれに,COL1A1における特定の病的変異は多発関節弛緩型EDS(OMIM 130060),EDS/OI重複表現型,または常染色体優性の幼児性皮質骨骨増殖症(caffey病)と関連している.
遺伝的に関連のある(対立遺伝子)疾患
SDHA
両アレルSDHA病原性バリアントは、遅発性の視神経萎縮症およびリー症候群(mtDNAバリアントによるリー症候群の考察については、ミトコンドリア障害の概要を参照)や、早期発症の進行性脳症を特徴とする神経変性疾患と関連している。
鑑別診断
易出血性,関節可動亢進,慢性的関節脱臼を伴っている場合,他のEhlers-Danlos症候群の病型について考慮すべきである.これらの病型は臨床所見が下記の点で古典型 EDSと重複している.
| 病型名 | 遺伝子 | 遺伝形式 | 臨床的特徴 | |
|---|---|---|---|---|
| cEDSとの共通点 | cEDSとの相違点 | |||
| 類古典型EDS (OMIM 606408) |
TNXB | 常染色体劣性 | ・易出血性 | |
| (OMIM 225320) | COL1A2 | 常染色体劣性 | ||
| 不明 | 常染色体優性 | ・軽度の萎縮性瘢痕 | ・紙状瘢痕および/または血管性瘢痕の所見がない | |
| 多発関節弛緩型EDS(OMIM 130060, 617821) | COL1A1 COL1A2 |
常染色体優性 | ・易出血性 | ・両側性先天性股関節脱 臼 |
| 皮膚脆弱型EDS (OMIM 225410) |
ADAMTS2 | 常染色体劣性 | ||
| 後側彎型EDS | PLOD1 | 常染色体劣性 | ・先天性筋萎縮症 | |
| 筋疾患,感音性難聴を伴う後側彎型EDS | FKBP14 | 常染色体劣性 | ||
臨床的マネジメント
合併症とマネジメントの詳細は,Bowen et al [2017]を参照のこと.
最初の診断時における評価
古典型EDS(cEDS)と診断された患者の疾患の程度を確定するために,以下が推奨される:
- 皮膚過伸展,萎縮性瘢痕や打撲傷の評価を加えた皮膚の臨床的観察と古典型 EDSの他の症状
- 関節可動性はBeighton 判定基準で判定する
- 幼児,小児期における筋緊張低下や運動発達を評価する
- 大動脈径の心エコーの基準値は診断時に測定する
- 重篤な易出血性がある場合は凝固因子を評価する
- 臨床遺伝専門医および/または遺伝カウンセラーとの連携
病変に対する治療
古典型EDSの小児で筋緊張低下や運動発達遅延を来している場合,理学療法プログラムは有用である.
体重負荷のない運動(例えば水泳)は筋肉の発達や協調を促す.
筋緊張低下や慢性的な疼痛を伴う関節不安定性のある方は,症状に合わせた生活様式に適応することが必要かもしれない.心理的支援,行動療法,心理療法は,受け入れの進行や対処能力に役立つ可能性がある.
皮膚創傷は張力をかけずに,通常2層に縫合する.全般に深い縫合を行うべきである.皮膚縫合は縫合領域を通常の2倍にし,瘢痕の伸展を防ぐために追加の固定やテープの使用が有用である.
出血時間を正常化するためには,DDAVPⓇ(酢酸デスモプレシン)が有用である.打撲や鼻出血,抜歯などの処置前などに有効である.関節弛緩や脱臼の治療に関する推奨事項は関節過可動型EDSを参照のこと. (注: 外科的な関節安定は効果が得られないか,一時的な改善に終わる).
抗炎症薬は関節痛を軽減することがある.
長期間の慢性的な痛みは,心理的支援が必要かもしれない.
心血管系の症状に対して,標準的な治療を行う.
関節弛緩および関節脱臼の初期症状の予防に関する推奨事項は,関節過可動型EDS:管理,初期症状の予防を参照.
アスコルビン酸(ビタミン C)は出血を軽減することがあるが,皮膚過伸展,萎縮性瘢痕,関節可動亢進の初期症状には効果がない.一般に,成人の摂取量は1日に2グラムが推奨されており,小児に対しては比例的に減量して投与する.ただし,制限はない.
二次病変の予防
関節弛緩および脱臼といった二次病変の予防については,関節過可動型EDS:管理,二次合併症の予防を参照.
経過観察
- 成人において心エコーで異常を認めない場合は,定期的な心エコーは必要ない. (大動脈拡張の進行に関する長期的なデータがなく,大動脈が正常径の人に対して,特別なフォローアップの推奨方法は有用ではない)
- 小児において心エコーで異常を認めない場合は,小児循環器専門医によるフォローアップが推奨される.
- 大動脈拡張や僧帽弁逸脱などの異常を認めた場合は,年1度の定期的な心エコーを行うべきである.
- 関節に負荷のかかるスポーツ(対戦相手がいるスポーツ,格闘技,サッカー,ランニング)
- アセチルサリチル酸(アスピリン)
リスクのある血縁者の検査
血縁者の内,一見無症候の高齢者や若年者の遺伝学的検査から病的変異を同定し遺伝的状態を明らかにすることは,早期治療や予防行動につなげる一助となる.遺伝カウンセリングとして扱われるリスクのある親族への検査に関する事柄は「遺伝カウンセリング」の項を参照のこと.
妊娠期の管理
会陰裂傷,出産後の出血,子宮脱,膀胱脱が起こるリスクが高いため,妊娠中および出産後のモニタリングが推奨される.
アスコルビン酸 (ビタミンC)が易出血性を軽減する可能性がある「一次病変の予防」を参照). 一般に,成人では1日2グラムの摂取が推奨されるが,妊娠後期の3ヵ月間の厳格な基準は存在しない.
早期破水のリスクが高い場合,早産のモニタリングは妥当である.
研究中の治療法
様々な疾患や病態の臨床試験に関する情報はアメリカのClinicalTrials.gov,欧州ではEUのClinical Trials Registerを参照.
注:この疾患に関する臨床試験が行われていない可能性がある.遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
古典型Ehlers-Danlos 症候群は常染色体優性遺伝形式をとる.
患者家族のリスク
発端者の両親
- 罹患者の約50%は罹患した片親から病的変異を伝達され,残りの約50%は新生突然変異で発症すると推測される.
- 一見したところ孤発例とみられる罹患者の両親に対して,創傷治癒の遅延,易出血性などの皮膚所見,関節可動亢進あるいは反復する脱臼や慢性の関節痛を評価すべきである.病的変異が発端者に特定された場合,両親の遺伝学的検査が推奨される.
- もし,親の白血球DNA中に病的変異が検出されない場合は,患者の新生突然変異,また親の性腺モザイクなどが考えられる.理論的にはありうるが,性腺モザイクの例は報告されていない.
- cEDSと診断された方の中には,家系内で疾患を認識していなかったり,理論的には浸透率が低下していたりするために,家族歴が陰性に見える人もいる(浸透率が100%か,あるいはそれより低いかは不明).したがって,一見陰性に見える家族歴は,適切な臨床評価および/または遺伝学的検査が行われない限り,正しいか確認することができない.
発端者の同胞
罹患者の同胞のリスクは親の遺伝的状況(罹患しているかどうか)による.
- 親が罹患している場合,どの同胞もリスクは50%である.家系内で表現型の違いがみられる.
- 同胞でみられた病的変異が親の白血球DNAから検出できない場合,親の性腺モザイクの可能性もあり,兄弟姉妹への再発リスクは1%と推定される[Rahbari et al 2016]
- 両親が病的変異同定の検査を受けていなくても,臨床的に症状がない場合,兄弟姉妹へのリスクは低いようである.
発端者の子
古典型 EDSの罹患者の子は50%の割合で病的変異を受け継ぐ.
家系内で臨床症状の違いがみられる.
発端者の他の家族
罹患者の他の家族に対するリスクは,親の遺伝的状況(罹患しているかどうか)による.親が罹患している場合,罹患者の他の家族もリスクを有する
遺伝カウンセリングに関連した問題
早期診断・早期治療を目的としたリスクのある親族の評価に関する情報については,「臨床的マネジメント リ
スクのある親族の検査」を参照.
リスクのある人
常染色体優性遺伝疾患では両親にいずれにも発端者に認めた特定の病的変異がなく,本疾患の症状がない場合,この病的変異は新生突然変異である可能性が高い.一方で,父あるいは母が違うとか(例えば生殖補助医療),公表していない養子縁組など,医学的でないことでもおこりうる.
家族計画
- 遺伝リスクの決定や出生前診断の利用について話し合う最適な時期は妊娠前である.
- 罹患している若年成人やリスクのある若年成人に対して遺伝カウンセリング(子への潜在的リスクや生殖手段についての話し合い)を行うことが適切である.
DNAバンキング
DNAバンクは主に白血球から調製したDNAを将来の使用のために保存しておくものである.検査法や遺伝子,変異,疾患に対する新たな知見が得られる可能性もあり,罹患者のDNAを保存することは考慮に値する.
出生前診断と着床前診断
疾患を引き起こす病的変異が同定されている罹患家族において,ハイリスク妊娠のための出生前診断,着床前診断が可能である.
特に遺伝学的検査が早期診断よりも中絶を目的として考慮される場合は,医療関係者と家族の間では出生前診断に対する見解の相違が生じるかもしれない.多くの医療機関では最終的には両親の意思を尊重するとしているが,この問題については注意深い検討が求められる.
訳注:一般に古典型 EDSに対して出生前診断の適応があるとは考えられておらず,日本では行われていない.
更新履歴
- Gene Review著者: Richard Wenstrup, MD Anne De Paepe, MD, PhD
日本語訳者: 訳者(所属) 渡邉 淳 (日本医科大学付属病院遺伝診療科)
Gene Review 最終更新日: 2006.4.10. 日本語訳最終更新日: 2007.4.4 - Gene Review著者: Richard Wenstrup, MD Anne De Paepe, MD, PhD
日本語訳者: 訳者(所属) 渡邉 淳 (日本医科大学付属病院遺伝診療科)
Gene Review 最終更新日: 2007.7.24. 日本語訳最終更新日: 2008.1.6 - Gene Review著者: Richard Wenstrup, MD Anne De Paepe, MD, PhD 日本語訳者: 吉村祐実(翻訳ボランティア) ,櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: 2011.5.29. 日本語訳最終更新日: 2016.4.11 - Gene Review著者: Fransiska Malfait, MD, PhD, Richard Wenstrup, MD, and Anne De Paepe, MD, PhD
日本語訳者: 関屋智子(金沢大学遺伝カウンセリングコース) ,渡邉淳(金沢大学附属病院遺伝診療部)
Gene Reviews 最終更新日: 2018.6.26 日本語訳最終更新日: 2020.7.19 (in present)
![]()