エマヌエル症候群
(Emanuel Syndrome)
GeneReview著者: Livija Medne, MS, CGC, Elaine H Zackai, MD, FACMG, Beverly S Emanuel, PhD, FACMG
日本語訳者:倉橋浩樹、大江瑞恵、細羽恵理子(藤田保健衛生大学・総合医科学研究所・分子遺伝学研究部門)
GeneReview最終更新日: 2010.5.11 日本語訳最終更新日:2010.7.20
要約
疾患の特徴
エマヌエル症候群は重度精神遅滞、小頭症、成長障害、前耳介小突起、前耳介洞、耳の奇形、口蓋裂または高アーチ状の口蓋、小顎症、腎異常、先天性心疾患、男性では生殖器の異常を呈する。
診断・検査
エマヌエル症候群は次のいずれかからなる染色体不均衡が原因となる。1)22番派生染色体[der(22)]を過剰染色体として持つ場合。核型は女性47,XX,+der(22)t(11;22)(q23;q11)、男性47,XY,+der(22)t(11;22)(q23;q11)。2)まれではあるが、均衡型t(11;22)(q23;q11)と過剰な派生染色体を持つ場合。過剰22番派生染色体は500-550バンドレベルの通常のG-バンド解析で容易に同定される。
臨床的マネジメント
対症療法:通常、多くの専門家チームによる介護が必要である。胃食道逆流症、鎖肛(また狭窄)、鼠径ヘルニア、心奇形、口蓋裂、先天性股関節脱臼、他の骨格合併症、難聴、停留睾丸および小陰茎症、眼の屈折異常、斜視や他の眼科学的な問題の標準的なマ ネ ジメント。
運動発達、職業や言語療法:コミュニケーションを促進させるための代替コミュニケーション法。二次的な合併症の予防:小児麻酔科医のいる病院で鎮静や手術の間、気道の状態に注意を払って行う。
サーベイランス:それぞれのケースにおいて全身の合併症の程度にもとづく、フォ ローアップが必要である。定期的な発達評価:臨床遺伝学者による定期的な再評価。
遺伝カウンセリング
99%以上の場合、エマヌエル症候群の発端者の両親のいずれかは t(11;22)(q23;q11) 均衡型保因者であり、表現型は正常である。ほとんどの場合、保因者である両親は、その親のいずれかから t(11;22)を受け継いでいる。両親のいずれかが保因者である発端者の同胞は、1)過剰22番派生染色体症候群、2)t(11;22)均衡型保因者、3)過剰22番派生染色体症候群、もしくは、他の減数分裂不分離による自然流産になる可能性がある。そのリスクは発端者の母か父のどちらが均衡型保因者であるかによって異なる。もし、その染色体異常が家族内で確かめられているのならば、リスクの高い妊娠のための出生前診断は可能である。
診断
臨床診断
エマヌエル症候群の特徴
- ●主要な表現型は下記に[Fraccaro ら1980 , Zackai & Emanuel 1980 , Lin 1986, Carterら2009]
- 重度精神発達遅滞
- 小頭症
- 成長障害
- 前耳介小突起、前耳介洞
- 耳の奇形
- 口蓋裂、高アーチ状口蓋
- 小顎症
- 腎奇形
- 先天性心疾患
- 男性の生殖器異常
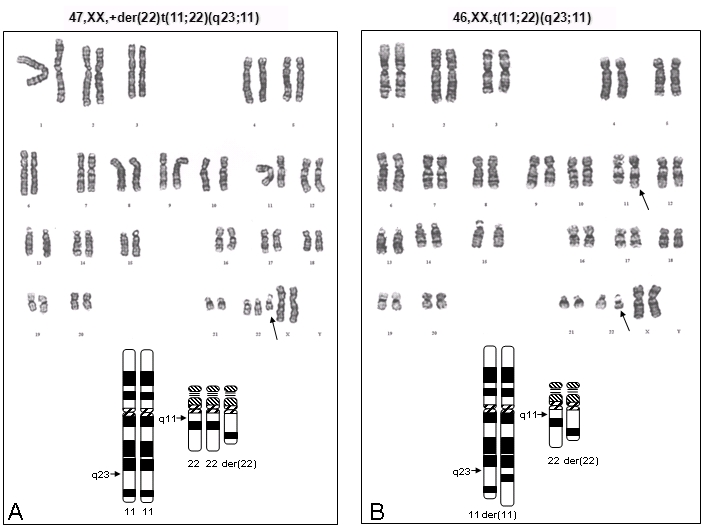
図1
A. 核型とイディオグラムの模式図は11q23-qter と22q cen-q11染色体のトリソミーとなる過剰22番派生染色体を示す。
B. 核型とイディオグラムの模式図は均衡型転座保因者を示す。
下記のいずれかからなる染色体不均衡(図1を参照)
- 通常、22番派生染色体[der(22) ](図1A)は下記の核型のように過剰染色体として持つ。
- 女性 47,XX,+der(22)t(11;22)(q23;q11)
- 男性 47,XY,+der(22)t(11;22)(q23;q11)
- 稀に、t(11;22)均衡型転座に加えて過剰派生染色体として持つ。図1Bに均衡型t(11;22)保因者の核型を示す。
検査
細胞遺伝学検査
染色体異常が疑われた時、通常の細胞遺伝学解析が推奨される。過剰 22番派生染色体は患者の500-550バンドの通常のGバンド解析によって容易に同定される。
- 両親のいずれかがt(11;22)均衡型転座保因者かどうかを決定するために、両親の核型を調べるべきである。
- 両親のいずれもが均衡型転座保因者でない稀な例では、22q11.2欠失検出用と11qのテロメア検出のための市販FISHプローブによって、核型の過剰な染色体が11番、22番染色体から由来していることを同定することができる。
これらの検査を組み合わせることにより、エマヌエル症候群の原因となっている小さな末端動原体型染色体を100%同定できる。
分子遺伝学検査
遺伝子
過剰22番派生染色体上の遺伝子の重複が、エマヌエル症候群と関係していることが知られている唯一の遺伝的欠損である。臨床症状は、過剰22番派生染色体上の11q23-qterの重複と22q10-22q11の重複に由来する。
注意: 22番染色体はヘテロクロマチンの短腕をもつ着糸点末端型染色体であるので、22番染色体の短腕の重複は、臨床的には意味はない。
臨床検査
- Gバンドによる染色体分析により過剰22番派生染色体を同定する。
- FISHテストは22q11.2に位置するN25またはTUPLE1プローブと11qサブテロメアプローブの使用により行われる。
- 11番と22番染色体プローブを使った全染色体ペインティング(WCP)により、過剰染色体が11番と22番に由来することを示すことができるだろう。この方法は通常は使用されず、FISHテストにより染色体解析で観察された過剰22番派生染色体の起源を容易に確認することができる。
- アレイゲノムハイブリダイゼーション(アレイCGH)は、動原体周辺、サブテロメア、22q11.2領域のバッククローンを含んだ全ての市販のBACアレイによって過剰染色体を検出する事ができる。加えて、オリゴヌクレオチドアレイは過剰der(22)による11qと22qの重複を検出する。
研究検査
- 切断点特異的PCRによる検査。切断点をまたいで増幅するプライマー対によるPCRは、典型的な反復性の転座切断点を確かめることができる[Kurahashiら2000a, ; Kurahashiら2000c]
- MLPA(multiplex ligation-dependent probe amplification)法は22qの重複を検出するために開発されてきた。MLPAはゲノムターゲット配列の相対的なコピー数を決定するための定量的マルチプレックスPCR法である[Vorstmanら 2006]
表1にエマヌエル症候群に使用できる分子遺伝学的検査の要約 を示す
| 検査法 | 検出する変異 | 変異検出頻度1 | 検査の利用 |
|---|---|---|---|
| 染色体解析 | 過剰22番派生染色体 | 100% | 臨床 |
| FISH | 22q11と11q23の重複 | 両方のプローブを使った場合100% | 臨床 |
| アレイCGH | 11番染色体と22番染色体のコピー数の変化 | 100% | 臨床 |
| MLPA | 22q11重複 | 100% | 臨床 |
検査の利用は、GeneTests Laboratory Directoryを参照している。GeneReviewsでは分子遺伝学的検査をUSCLIAで承認された研究所もしくはUS以外の臨床検査所によって、GeneTests Laboratory Directoryにリストされた検査を臨床利用として指定している。GeneTestsは研究所が提出した情報や検査所のライセンスやパフォーマンスなどすべて保証しない。臨床家は情報を確かめるために研究所に直接連絡をしなければならない。
- 示された遺伝子中に存在する変異を検出する検査方法の能力。
検査手順
- 発端者の診断の確定や確認。過剰22番派生染色体が疑われた時、過剰染色体は過剰にある小さな着糸点末端型染色体として簡単に同定されるので、通常の細胞遺伝学的分析を行う。
- 過剰の染色体が一部は22番染色体から由来したことを判定するために、22qのFISHプローブを使用する。
- 転座が11qと22qの間であることを確かめるために、11qテロメアプローブを使用する。
- リスクのある血縁者のための保因者診断には、前もって家族メンバーの転座の同定が必要である。(均衡型転座保因者は典型的には無症状である。)
- リスクのある妊娠のための出生前診断には、前もって家族内の転座の同定が必要である
遺伝的に関係のある疾患
過剰22番派生染色体と関係のある他の表現型はない。
t(11;22)均衡型保因者女性は、閉経前の乳癌のリスクがいくらか増加するかもしれない。この関連は初めにLindblomらによって提案された(1994)。彼らは8家系の5家系中には1人の乳癌の女性がいることを報告した。さらに最近、2つの無関係の家系で、何世代かにわたって、何人かで乳癌が均衡型t(11;22)と一緒に遺伝した例を報告した[Jopanputra ら2005, Wieland ら2006)。しかしながら、たくさんの家系をみている他の研究では、この関連は確認されていない[Kurahashi ら2000c]。
80家系を調査した最近の研究での結論は、(11;22)均衡転座保因者の乳がん発生率は、一般人以上に増加していない。 [Carter ら 2010]。研究結果は、もし乳がんの家族歴がある場合に限って、t(11;22)女性均衡転座保因者は乳がん健診をしっかりすることが推奨されるということを示唆している。さらに、この研究では、(11:22)均衡転座の保因者の食道癌や黒色腫の危険性の増加は否定できなかった。この問題を解決するためには、さらに大規模な研究が必要である。また、これらの結果の結論として、(11:22)均衡転座の保因者は日焼け防止を強化し、皮膚病変をチェックし、一般集団のように癌の可能性のある兆候に細心の注意を払うことを推奨する。臨床像
自然経過
100名以上の過剰22番派生染色体症候群の報告がなされている
[Fraccaro ら 1980 , Zackai & Emanuel 1980 , Lin ら 1986]。先天性心疾患、横隔膜ヘルニア、腎不全といった重篤な先天性奇形は主な死亡原因となる。一番死亡率の高いのは、生後一ヶ月である。対症療法の改善や時間経過により、生存の機会が改善され、成人期までの生存がよく報告されるようになってきた。
患児は、通常新生児期に、t(11;22)の均衡型転座保因者の子孫としてみつかる。
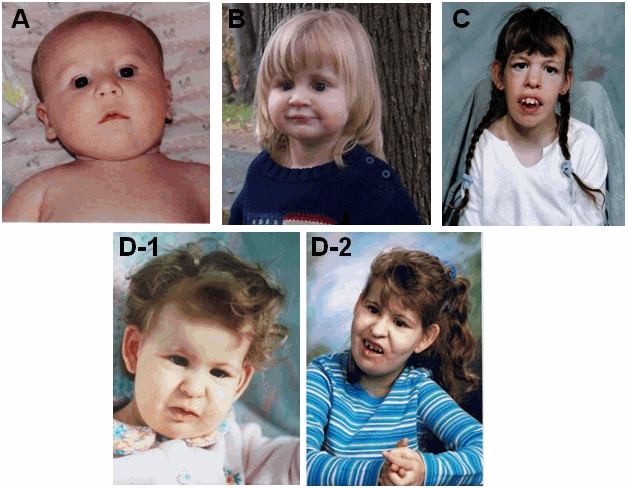
図2 写真
エマヌエル症候群の4人。
患児の丸顔や、深く窪んだ丸い目や、前額の突出に注意。A(生後6ヶ月以内)、B(3才)。
時間経過に伴い顔貌が粗野になってくるのに注意 D。
D-1は1才、D-2は10才での写真。
顔貌の特徴は、年齢が増した2人の患者の間で非常に似ている。C(17才)とD-2。
体格
ほとんどの患者は、出生前、出生後の成長遅延と、関連した形態異常を呈する。
頭部・顔面
顔面の形成異常は、小短頭症、前額突出、瞼鼻ひだ、垂れ下がった眼裂、広く平坦な鼻梁、長くてはっきりとした人中、小下顎症と下顎後退、耳介前部の小孔や小突起としばしば関連した耳介異常(小耳症から大きな耳まで)を呈する。(図2をみよ)
心臓
先天性心疾患は過剰22番派生染色体症候群の約60%でみられ、死亡率に寄与している。
心奇形には、心房中隔欠損症、心室中隔欠損症、ファロー四徴症、総動脈幹、三尖弁閉鎖症、大動脈縮窄症、鎖骨下動脈走行異常、左上大静脈遺残や動脈管開存症がある。
泌尿生殖器
腎奇形は、患者の約30%にみられ、完全な先天性腎無形成からさまざまな程度の腎形成不全までみられる。男性はしばしば停留睾丸、小陰嚢、小陰茎症をともなう性器発育不全を呈する。女性は、子宮奇形が時折観察される。
胃腸
横隔膜ヘルニアと形成不全、または、横隔膜弛緩症がみられた。
瘻孔の有無にかかわらず、鎖肛症は患者の約20%が認められる。完全な閉鎖のない肛門狭窄症も、よくみられる。
鼠径ヘルニアは、頻度は高くないがよく記載されている。
胆道閉鎖症、ヒルシュスプリング病、肝分葉異常、肝外胆管、胆嚢欠損、多脾症などは、時折みられる。
体重増加不良はよくみられる。特徴的な摂食問題はあまり報告されていないが、胃食道逆流や吸乳や嚥下の困難もよくみられる。
筋骨格
患者は、著しい中枢性の筋緊張低下を呈する。
脊柱弯曲症は、重症な筋緊張低下や運動遅延の二次的な合併症としておこっている可能性が高い。
先天性股関節脱臼もしくは亜脱臼はよくみられる。
クモ指症や先が細い指は特徴的である。
内反足や関節拘縮は、先天性もしくは後天性に発症する。
他にも、それほど多くはないが13対の肋骨や鎖骨形成不全、外反射、橈骨尺骨融合や足趾の4-5合趾症などといった骨格形成異常がみられる。
腰部脊髄髄膜瘤が一度報告されている。仙骨のくぼみは、よくみられる。
骨年齢の遅延が、少数の症例報告で述べられている。
口蓋
口蓋裂は、患者の約50%でみられる。
目
エマヌエル症候群のほとんどの患者の視力は正常である。多くはないが、斜視や近視といった眼異常を呈する。眼瞼下垂症や網膜変性症はさらにまれである。
耳・鼻・咽頭
外耳介の奇形は典型的で、耳介の前の小孔や小突起が特徴的である。外耳道閉鎖や難聴を伴う重度の小耳症が報告されている。聴覚障害の頻度は高くないが、重度の発達障害により正確な聴力評価が難しいので、軽度のものは見逃されているかもしれない。
とがった口角や亀裂、上顎裂、喉頭軟化症、そして鰓溝遺残が報告されている。二分口蓋垂も呈する。中枢神経系
小頭症は、全ての患者でみられる。
脳の構造的な奇形の発生率は、診断を確定するために脳画像診断を必要としないので解っていない。報告された奇形はダンディーウォーカー奇形、脳梁欠損症、無嗅脳症や嗅球や嗅索の欠如である。
てんかん発作は、少しの患者で報告されており、別の少数の患者で臨床的なてんかん発作のない脳波異常が報告されている
発達
エマヌエル症候群の全ての患児は重度の全身の発達遅延を呈し、最重度の精神発達遅滞を呈する。ほとんどの患者は無支持で座ることができる.協調運動が欠如しているので、歩行はしばしば困難である。よってほんの少しの患者だけしか歩行を獲得することができない。発語と言語発達は、大きく遅延する。言葉の理解は、発語よりよく、何人かはコミュニケーション時に1語を使うことができる。その他
先天性免疫グロブリン欠損症、胸腺依存性免疫不全症、歯の形成異常。
遺伝型と臨床型の関連
エマヌエル症候群のすべての患者は過剰22番派生染色体をもつが、11q23と22q11上の切断点はほとんど一致している。切断点は数塩基のみ異なる[Shaikh ら 1999 ; Kurahashi ら 2000b ; Kurahashi & Emanuel 2001]。しかし、遺伝型と表現型の相関は、臨床所見が重複遺伝子に起因するのでむずかしい。全身的な合併症は多様である一方、発達の転帰は最重度精神発達遅滞という重症度では一様である。
浸透率
浸透率は過剰22番派生染色体症候群の患者では完全である。
促進現象
促進現象は過剰22番派生染色体症候群の患者に無関係である。
病名
過剰22番派生染色体症候群は、たいてい罹患していない親の均衡型t(11;22)の3:1不分離により生じる「Shaikh ら1999」。
Gバンドができる前の古い症例報告では、“部分トリソミー22”や“部分トリソミー11”としてこの染色体異常が報告されていた 。
2004年に過剰22番派生染色体症候群は、エマヌエル症候群と命名された。
有病率
過剰22番派生染色体症候群はまれな染色体異常であり、有病率は解っていない。一般集団中の均衡型t(11;22)保因者の頻度は解っていない。
鑑別診断
エマヌエル症候群と重なっている臨床的特徴は下記の症候群でみられる。染色体解析によってエマヌエル症候群の診断は確定し、他の疾患と鑑別診断される。
- Fryns 症候群
- Smith-Lemli-Opitz 症候群
- Pallister-Killian症候群
- Kabuki 症候群
- Wolf-Hischhorn 症候群
- 他の染色体異常
臨床的マネジメント
初期診断後の評価
臨床症状の出現率や死亡率への寄与を評価するようなガイドラインは現在までに報告されていない。エマヌエル症候群と診断された患者の疾患の程度を決定するための下記の提案は文献や著者たちの経験に基づくものである。
- 心奇形をスクリーニングするための心エコーでの心機能評価。心房中隔欠損症は、最も頻度が高いが、聴診だけでは検出することができないかもしれない。
- 腎臓形態異常の評価には、腎臓超音波検査。もし適応があれば、膀胱尿管逆流症の評価のために排尿時膀胱尿道造影
- 口蓋裂のために口蓋の評価
- 胃腸管の構造異常のための適切な放射線検査による胃腸管の評価。とくに、肛門狭窄症、横隔膜異常、胃食道逆流症の。
- 摂食や嚥下の評価
- 股関節異形成、ほか、関節性拘縮や内反足、脊柱彎曲、撓尺骨癒合症のための適切な放射線検査による整形外科的評価
- 外耳道の狭窄症もしくは閉鎖症のための耳鼻咽喉科の評価
- 聴覚脳幹性検査と耳音響放射検査による聴能学評価 (この検査についてのさらなる情報は、Hereditary Hearing LossもしくはDeafness を参照)
- 拡張型眼底鏡検査、視力の査定や斜視の評価のために眼科的評価
- 停留睾丸および小陰茎症の男性での泌尿器科の評価
- コミュニュケーションスキルに重点をおいた教育や治療的介入を行う小児発達医や療法士による評価
- 臨床遺伝カウンセリングや、リスクのある血縁者の同定のための遺伝学的評価。(過剰22番派生染色体はほとんど保因者である両親のひとりから受け継がれている。)
病変に対する治療
エマヌエル症候群の患者の年齢や全身合併症の程度によっては、多方面の専門領域からの健康管理プロバイダーによる評価が必要である。
いくつかの例では、重篤な構造異常や腎不全がある時、対症療法が必要である。- 胃食道逆流の標準な治療。もし成長障害があった時には、添加ミルクや経腸栄養の考慮
- 鎖肛(や狭窄症、もし適応があれば)や鼠径ヘルニアのための外科的矯正
- 標準な治療法
- 心奇形
- 口蓋裂
- 股間節異形成や他の骨格合併症。移動のためにしばしば必要とされる歩行器のような補助器具
- 難聴
- 停留睾丸および小陰茎症
- 屈折障害や斜視、もしくは他の眼科的問題
- てんかん発作 もしあれば
- 発達の結果を最適化するための運動機能、職業や言語療法
- 言語スキルはしばしば限界があるので、コミュニュケーションを促進するための代替コミュニュケーション法
二次病変の防止
鎮静や手術の中のケアは、エマヌエル症候群の患児は気道が狭く、多彩な口蓋の異常、喉頭軟化症がみられるので小児麻酔科医が携わる。
定期検査
以下のようにすることが、適切である。
- 患者の全身的な合併症の程度に基づいたフォローアップが必要とされる。
- 治療的介入や教育を容易にするために発達段階の定期的な評価
- 家族に新しい情報もしくは提案を知らせるための遺伝医学者による定期的な再評価
リスクのある血縁者の検査
遺伝カウンセリング目的のためのリスクのある血縁者の検査に関する問題点に関しては遺伝カウンセリングを参照
研究レベルの治療
広範囲に渡る病気や症状に対する臨床的研究の情報へのアクセスは、ClinicalTrials.gov.で検索する。
注意:この疾患の臨床治験はないかもしれない。
その他
患者やその家族は自然歴、治療、遺伝様式、他の家族への遺伝的リスク、生活用品に関して情報をもつべきである。
遺伝学の専門家による遺伝クリニックは、自然歴、治療、遺伝様式、他の家族への遺伝的リスク、そしてまた、利用できる生活用品に関して、患者やその家族の情報源である。
GeneTest のClinic Directoryを参照。
この疾患の包括的もしくは疾患に特異的はなサポート組織についてはConsumer Resourcesを参照。これらの組織は、他の患者との交流、サポート、情報提供をするために患者と家族のために設立されている。
訳注:海外のサポートグループに関する情報は、以下のウエブを参照。
「22番染色体セントラル」 http://www.c22c.org/
現在、日本では本症に対するサポートグループはないが、設立の計画はある。
情報源としては、以下のウエブがある。
「t(11;22)」 http://www.fujita-hu.ac.jp/~genome/11&22/
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
エマヌエル症候群は遺伝性の染色体異常である。t(11;22)(q23;q11)均衡型転座保因者の減数分裂時の3:1分離によりおこる。この転座はヒトで唯一、繰り返しおこることが知られている非ロバートソン転座である。
血縁者のリスク
発端者の両親
- 99%以上の場合、エマヌエル症候群の発端者の両親のひとりはt(11;22)(q23;q11)均衡型転座保因者で、表現型は正常である。父方の生殖細胞で発生した新生転座t(11;22) の、おそらく、不均衡な隣接I型分離と、母方の第一減数分裂時での22番染色体の不分離により発生した過剰22番派生染色体症候群の1例の報告がある。
- エマヌエル症候群の発端者の両親は染色体検査を提案されるべきであろう。
- 統計的には、過剰22番派生染色体を持つ発端者の母は、父よりt(11;22)均衡型保因者である可能性が高い。
- ほとんどの場合、保因者である両親のひとりは、その両親の片方からt(11;22)を受け継いでいる。
発端者の同胞
エマヌエル症候群の所見がない発端者の同胞
- エマヌエル症候群の所見がない発端者の同胞
- エマヌエル症候群のリスクはない。
- 別の不均衡な染色体異常をもっている可能性はほとんどない。
- 均衡型転座をもっている可能性は50%と推定される(表2)。t(11;22)均衡型転座保因者と同定された同胞の子孫は、以下に述べられるリスクをもつ(両親の片方が…を参照)。
- 正常な染色体をもつ可能性は50%と推定される。
- エマヌエル症候群の所見(例えば、重症な発達遅延、発育不良、多発性先天性奇形)をもつ発端者の同胞は、ほとんど過剰22番派生染色体をもっている。
- 発端者の両親の片方がt(11;22)均衡型保因者であれば、両親の将来の妊娠は以下のうちの1つになるリスクが増加する。
- 過剰22番派生染色体症候群
- t(11;22)均衡型保因者
- 過剰22番派生染色体や他の減数分裂時の不分離 による自然流産
- 表2のリスクは家系の生殖歴に基づいている。リスクは発端者の母か父のどちらが均衡型転座保因者であるかによって変わる[Fraccaroら 1980,Zackai&Emanuel 1980]。
t(11;22)均衡型転座保因者の子孫 表2を参照
表2 t(11;22)均衡型転座保因者の妊娠の結果
| 女性保因者 | 男性保因者 | 全体 | |
|---|---|---|---|
| 過剰22番派生染色体の生児 | 5.7%-6.1% | 2.2%-5% | 1.8%-5.6% |
| 均衡型t(11;22)の生児 | 55.4% | 41.2% | |
| 自然流産 | 23%-37% | ||
発端者の子孫
エマヌエル症候群の患者は重度の認知障害を有するために子孫をえる可能性は低い。
発端者の他の家族
他の家族のリスクは発端者の両親の状態に依存する。両親がt(11;22) 均衡型保因者だった場合、その家族はエマヌエル症候群か保因者のリスクがある。保因者であると同定された場合は上記に述べたのと同じリスクである(表2)。染色体分析はリスクのある家族に提案されるべきである。
保因者診断
リスクのある家族は染色体分析によって検査できる。保因者検査はリスクのある家族が未成年者の場合は通常行わず、成人で均衡型転座保因者であることの生殖に関する意味を理解できる場合に行われる。
発症していない同胞は通常、法律上成人となり、生殖年齢に達し、均衡型転座保因者であることの生殖に関する意味を理解することができる時に、保因者かどうかを検査する。遺伝カウンセリングに関連した問題
家族計画
- 遺伝学的リスク評価や出生前検査の可否などについての議論は妊娠前に行うのが望ましい。
- 均衡型転座保因者であったり、そのリスクのある若年成人に遺伝カウンセリングを提案することは妥当である。(子孫へのリスクの可能性や、生殖に関する選択枝の議論を含めて)。
出生前検査
リスクのある妊娠について出生前診断が技術的に可能である。 DNAは胎生約16-18週に採取した羊水中細胞や約10-12週*に採取した絨毛から調製する。検査のために組織を得る2つの方法では、過剰22番派生染色体を検出する感度は同等である。
*胎生週数は最終月経の開始日あるいは超音波検査による測定に基づいて計算される。
着床前検査
着床前診断はいくつかの事例で行われ、成功しており、リスクの高い家族のために利用できるかもしれない[Van Assche ら 1999]。PGDを提供している施設に関してはtestingをみよ。
*GeneTests Laboratory Directory中にリストされた研究所から利用できる検査の臨床使用を盛り込むことは、GeneReviews のポリシーである。審査者、編集者、著者によるそのような使用の承認を言及する必要はない。
分子遺伝学
表B.エマヌエル症候群のOMIMエントリー
| 609029 | EMANUEL SYNDROME |
分子遺伝学的発症機序
エマヌエル症候群は多くの遺伝子を含んでいる11qと22q染色体の重複したゲノム領域からなるので、分子遺伝学的発症機序は解らない。
謝辞
私達の研究活動にご協力頂いた、ステファニー・セントピエールさん、全てのご家族の皆様、「22番染色体セントラル」に感謝いたします。
更新履歴
- GeneReview著者: Livija Medne, MS, CGC, Elaine H Zackai, MD, FACMG, Beverly S Emanuel, PhD, FACMG
日本語訳者:倉橋浩樹、大江瑞恵、細羽恵理子(藤田保健衛生大学・総合医科学研究所・分子遺伝学研究部門)
GeneReview最終更新日: 2007.4.20. 日本語訳最終更新日: 2008.1.30. - GeneReview著者: Livija Medne, MS, CGC, Elaine H Zackai, MD, FACMG, Beverly S Emanuel, PhD, FACMG
日本語訳者:倉橋浩樹、大江瑞恵、細羽恵理子(藤田保健衛生大学・総合医科学研究所・分子遺伝学研究部門)
GeneReview最終更新日: 2010.5.11 日本語訳最終更新日:2010.7.20 (in present)