低ホスファターゼ症
(Hypophosphatasia)
Gene Review著者: Etienne Mornet, PhD, Mark E Nunes, MD
日本語訳者:渡邉 淳 (日本医科大学付属病院遺伝診療科)
Gene Review 最終更新日: 2016.2.4. 日本語訳最終更新日: 2016.5.12.
原文: Hypophosphatasia
要約
疾患の特徴
低ホスファターゼ症は,血清と骨アルカリホスファターゼの活性低下により,骨ならびに/あるいは歯に生じる不完全な石灰化を特徴とする.臨床症状は,胎児期から骨の石灰化がみられず死産となる重症例から,成人後期で生じる下腿の病的骨折という軽症例にまで及び幅が広い.低ホスファターゼ症で重症度は連続的なスペクトルとして表されるが, 診断時の年齢や重症度により通常以下の6つの臨床病型に分類されている
- 周産期重症型低ホスファターゼ症は,呼吸不全および高Ca血症によって特徴づけられる,
- 周産期良性型低ホスファターゼ症では,骨格の症状は胎児期から有するものの,症状はゆっくりと軽症の病型に移行する,
- 乳児型低ホスファターゼ症は,出生時から6ヶ月の間に血清アルカリホスファターゼ活性が上昇せずくる病の病像を伴う,
- 小児型低ホスファターゼ症は,骨密度が年齢相当より低下し,原因不明の骨折から,くる病の症状とともに乳歯の早期脱落を来す,
- 成人型低ホスファターゼ症は,成人期に下肢の疲労骨折・偽骨折により特徴づけられ,時に歯の早期脱落を来す,
- 歯限局型低ホスファターゼ症は,骨格症状はなく,乳歯の早期脱落および/または重症の虫歯という歯の症状のみを認める
診断・検査
正式な診断基準は確立されていないが,低ホスファターゼ症(偽性低ホスファターゼ症を除き)のすべての病型は共通して未分画血清アルカリホスファターゼ(ALP)活性の減少,ならびにアルカリホスファターゼの組織非特異的アイソザイム(TNSALP)をコードするALPL遺伝子に1つあるいは2つの病的変異が存在する.
臨床的マネジメント
症状に対する治療:
周産期型(重症型): 経験は少ないが期待されている,酵素補充療法 (ERT)や家族への支援. 乳児型や小児型の早期: 酵素補充療法(アスホターゼ アルファ), 呼吸管理,高カルシウム血症/尿症の治療.痙攣発作に対するビ タミンB6治療,頭蓋骨癒合症への治療.他のすべての病型:1歳から開始する定期的な歯科診療,変形性関節症,骨痛および骨軟化症に対する非ステロイド性抗炎症薬(NSAIDs,偽骨折および疲労骨折に対する内固定術.
サーベイランス:
1歳から年2回の歯科検診,乳児型の小児では頭蓋骨癒合症により二次的に生ずる頭蓋内圧亢進のモニタリング.
回避するべき薬剤/状況:
ビスフォスフォネート,過剰のビタミンD.
遺伝カウンセリング
周産期型および多くの乳児型低ホスファターゼ症は常染色体劣性形式を示す.軽症の病型(特に成人型および歯限局型低ホスファターゼ症)では,ALPL病的変異によるTNSALP活性の影響により,常染色体劣性または常染色体優性遺伝形式を示す.
- 常染色体劣性形式を呈する低ホスファターゼ症では,ヘテロ接合(保因者;キャリア)は変異部位により生化学的異常のみで臨床的に異常はなく症状を示さない者から,軽い症状を呈する者までがいる.新生突然変異の報告はあるものの,多くの場合罹患者の兄弟は25%の割合で罹患し,50%の割合で「無症候性の」保因者,そして25%の割合で保因者ではない非罹患者となる.
- 常染色体優性形式を呈する低ホスファターゼ症では,すべての発端者は罹患した片親をもち,これまでに新生突然変異は報告されていない.常染色体優性低ホスファターゼ症の罹患者の子は50%の割合で遺伝子変異を受け継ぐ.
妊娠時における出生前診断は,リスクが想定されていて,家系内での罹患者の病的変異が明らかな場合のみ行うことができる.周産期型ならびに乳児型低ホスファターゼ症の再発は出生前の超音波検査で確認できることがある.
低ホスファターゼ症は,
- 骨ならびに/あるいは歯の石灰化の不完全
- 病変のない歯根を有する乳歯の早期脱落
- 血清 アルカリホスファターゼ(ALP)活性低値 を有するとき疑われるべきである.
診断時の年齢や重症度により,現在少なくとも6つの臨床病型が分類されている(表1).
臨床的特徴を下記に挙げる:
- 出生前からの長管骨屈曲:骨軟骨性棘および脛骨前部のえくぼ徴候を伴う
- 清アルカリホスファターゼ活性が上昇しない幼児のくる病病変: 成長障害,頭蓋癆,頭蓋骨癒合症,青色強膜,肋軟骨の拡大(「肋骨念珠」),側弯,手首や足首の厚み,長管骨屈曲,弛緩した靭帯および筋緊張低下も含む.
- 高カルシウム血症および高カルシウム尿症:生後1歳までに認める
- 病的骨折および骨痛:成長期の小児の骨折は骨幹端に生じる傾向がある.しかし,骨折は骨端や骨幹も認める.成人では,中足骨骨折および大腿骨の偽骨折を起こしやすい.
- 切歯から始まる乳歯の早期脱落:脱落歯には歯根が残っており,他の疾患ではみられなくなく本症に特徴的である.永久歯の虫歯および早期脱落あるいは抜歯もみられる.
- 家族歴:どの病型の低ホスファターゼ症においても症状の程度は様々であるが,常染色体劣性遺伝形式あるいは常染色体優性遺伝形式と一致する.
低ホスファターゼ症のX線像:年齢および病型に応じて様々であり,特徴的である. 周産期型(致死型)低ホスファターゼ症はX線像で明確となる.軽度な症例では,X線像が特徴的ではないため,臨床像,検査結果およびX線像の組み合わせが診断に必要となる.
- 骨減少症,骨粗鬆症,あるいは年齢に比した低骨塩量:二重エネルギーX線吸収測定法(DEXA)によって同定できる.骨塩量が加齢に伴って増加し,思春期では改善するものの中年時に再発することがある.
- 幼児くる病:石灰化が不十分な骨,広くみえる縫合線,短頭,胸郭動揺(フレイルチェスト),くる病性肋軟骨変化(参照 図 1A),広がった骨幹端(手首,膝および足首の広がりにつながる),骨化の乏しい骨端骨,屈曲した足という所見を含む.
- 乳歯の早期脱落による歯槽骨の消失:多くは下顎骨前部に起こり,まず中切歯が最初に失われる.しかし,いかなる歯も影響を受ける (参照 図1B).
- 骨幹端に限局的な骨欠損:X線透過性で「舌」のように見える.(参照 図1C). この所見は,小児型低ホスファターゼ症に特徴的である.
- 中足骨の疲労骨折:小児型と成人型低ホスファターゼ症にみられる
- 側面の偽骨折(骨改構層)を伴う骨軟化症:成人型低ホスファターゼ症に見られる(参照 図1D).
図1 低フォスファターゼ症の画像的特徴
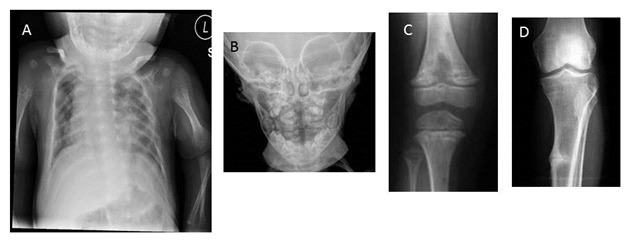
A. 乳児型低ホスファターゼ症における,くる病性肋軟骨変化, 胸郭動揺(フレイルチェスト), 骨幹端形成異常(近位上腕骨)
B. 小児型低ホスファターゼ症における,臼歯を囲んだ歯槽骨損失
C. 小児型低ホスファターゼ症におけるX線透過性で「舌」のように見える骨幹端
D. 成人型低ホスファターゼ症における骨改構層 (偽骨折)
表1病型による低ホスファターゼ症の臨床像
| 病型 | 遺伝形式1 | 臨床像 | 歯病変 | 臨床診断 |
|---|---|---|---|---|
| 周産期型 (重症型) | AR | 低石灰化,骨軟骨性棘突起 | ±2 | 画像診断,出生前超音波検査 |
| 周産期型(良性) | AR or AD | 長幹骨屈曲,出生後の良性経過 | ± | 出生前超音波検査,臨床経過 |
| 乳児型3 | 多くは AR | 頭蓋骨癒合症,低石灰化,幼児くる病,高カルシウム尿症 | 乳歯の早期脱落 | 臨床経過,画像診断,検査所見 |
| 小児型 | AR or AD | 低身長,骨格異常,骨痛,/骨折 | 乳歯の早期脱落, (切歯) |
臨床経過,画像診断,検査所見 |
| 成人型4 | AR or AD | 疲労骨折: 中足骨,頸骨, tibia; 軟骨石灰化症 | ± | 臨床経過,画像診断,検査所見 |
| 歯限局型 | AR or AD | 歯槽骨喪失 | 抜歯 (切歯), 虫歯 | 臨床経過,歯科パノラマ撮影,検査所見 |
- AR = 常染色体劣性; AD =常染色体優性
- 以前は,重症の病型の多くは,(歯の) 萌出(ほうしゆつ)や脱落する前に死亡した.周産期型 (重症型) や 乳児型が治療できる現在,歯の所見はまだ明確に判っていないが,今後示されるだろう.
- 血清ALP活性が正常である乳児低ホスファターゼ症はほとんど報告がなく,偽性低ホスファターゼ症と定義している.偽性低ホスファターゼ症の生化学的かつ分子基盤は明らかではない.
- 成人型低ホスファターゼ症の方は,通常小児型,乳児型および周産期型の低ホスファターゼ症の既往を有することがある.
検査
全血清アルカリホスファターゼ(ALP)活性:低値.低ホスファターゼ症のすべての病型で,血清ALP活性は低値となる.
- 国内外の検査室ごとに異なる方法で測定され,基準値の範囲も異なる.それぞれの検査室で決定された性別・年齢別基準基準値を使用すべきである. 参照 表2に主要な検査室における基準値の下限値
- を示す.
- 罹患者でも妊娠中に血清ALP活性が一時的に増加する.血清ALP活性は,肝臓疾患,骨折急性期あるいは手術時に微増する.従って幼児期に原因不明の骨折により本疾患を疑う際には,血清ALP活性の経時的な測定が必要となる.
- 血清ALPアイソザイムによる骨分画活性の定量は通常必要ではない.しかしながら,肝疾患を合併している場合,血清ALP活性は「偽って」正常かもしれない.骨分画は熱に不安定だが,肝分画は熱に安定である.
訳注)
日本のALPの基準値は下記に示す北アメリカ(カナダ)とは,単位や測定法が異なり,各施設の年齢別基準値(下限値)があればそれを基に判定する.指定難病「低ホスファターゼ症」診断基準では,成長期の小児では血清ALP値が300IU/L未満では、本症を疑う必要があると述べられている.
表2 北アメリカにおける血清アルカリフォスファターゼ活性のための典型的な正常な基準最低値
| 血清アルカリホスファターゼ活性正常下限値(U / L) | ||
|---|---|---|
| 男 | 女 | |
| 0-30 日 | 60 | 60 |
| 1-11 月 | 70 | 70 |
| 1-3 歳 | 125 | 125 |
| 4-11 歳 | 150 | 150 |
| 12-13 歳 | 160 | 110 |
| 14-15 歳 | 130 | 55 |
| 16-19 歳 | 60 | 40 |
| 20 歳 | 40 | 40 |
注
(1) 正常値は使用した手法およびサンプリングされた集団によって異なる.全血清または血漿アルカリホスファターゼ活性の正常下限値は,測定した検査室に特有であり,検査室間で異なる.
(2) 検査室での経験を考慮した基準値を優先的に使用すべきである.
(3) 小児期の基準値の年齢間隔はカナダ人民族を用いて提案した[Colantonio et al 2012] .
尿中ホスホエタノールアミン(PEA)濃度:上昇.
- 低ホスファターゼ症において最も一般に行われる二次スクリーニング法である.尿中アミノ酸分析の一部として得られる.
- 尿中PEA濃度高値は,低ホスファターゼ症の診断の助けになる.しかし,尿中濃度は,他の代謝性骨疾患でも増加することもあり,罹患者でも正常である場合もある.
注:尿プロリン濃度の上昇は,検査結果の特異性を高める. - 無症候性保因者(ヘテロ接合体)はときに血清ALP活性が低下し,尿中PEA濃度が上昇している.
血清ピリドキサール5'-リン酸(PLP)濃度:高値.
- ビタミンB6の生理活性代謝産物であるPLP値は低ホスファターゼ症の最も感度の高い指標かもしれない[Cole et al 1986] .
- タミン B6を測定する多くの基幹検査所では,PLPを測定し,(1)ビタミン B6あるいは (2) PLP 値として報告している.従って,PLP の選択肢がなくてもビタミン B6の検査依頼で十分かもしれない.
- 検査前一週間以内にビタミンサプリメントを使用した場合,血清PLP濃度検査は偽陽性を示すことがある.
血清カルシウム,イオン化カルシウムおよび無機リン酸濃度:正常
- 本検査正常値は,低ホスファターゼ症と他のくる病を区別する.
- 高カルシウム尿は,血清カルシウム濃度の高値の有無に関わらずおこりえる.
- 血清または尿中無機リン酸濃度は,正常域が最も典型的であるが,高値を示すこともあり,幅が大きく診断に用いることは難しい.
25-(OH) ビタミンD,また1,25-(OH)2ビタミンDおよび副甲状腺ホルモン(nPTH)の血清濃度:正常
尿無機ピロリン酸塩(PPi):上昇
- 罹患者および無症候性ヘテロ接合体において感受性が高いマーカーである.
確定診断に向けて
遺伝学的検査が主体となる出生前期を除けば,低ホスファターゼ症の多くはルーチンに行う臨床生化学検査や画像で診断できる.確定診断は,発端者 の ALPL分子遺伝学的検査で両方のアレルあるいはヘテロで病的変異の同定による.参照 表 1).分子遺伝学検査は,単一遺伝子検査,多遺伝子パネルから包括的な検査まである.
- ALPL 遺伝子の単一遺伝子検査: 塩基配列解析を最初に行い,病的変異が1アレルの同定あるいは同定できない際には,続けて標的遺伝子の欠失/重複解析を行う.
- ALPL を含む関連遺伝子を搭載した多遺伝子パネル: (参照 鑑別診断) (注意)パネルに搭載される遺伝子や遺伝子ごとの診断の感度は診断施設により幅がある.
- 全エクソーム解析 (WES)や全ゲノム解析 (WGS)を含んだ網羅的遺伝学的検査: 低ホスファターゼ症の臨床像を有するのにALPL遺伝子を含む単一遺伝子検査や多遺伝子パネルで変異を同定できないときに考慮される.このような検査の供給や提案は以前には考慮されなかった(すなわち,類似な臨床症状で異なる遺伝子の変異を有する)
表3 低ホスファターゼ症に使用される分子遺伝学的検査
| 遺伝子1 | 検査手法 | 発端者における病的変異2の検出率 |
|---|---|---|
| ALPL | 塩基配列解析3 | ≈95%4,5 |
| 標的遺伝子欠失/重複6 | 不明7 |
- 染色体座位とタンパクは,表 A. 遺伝子 とデータベースを参照. .
- この遺伝子の変異情報は,分子遺伝学を参照.
- 塩基配列解析により良性, おおよそ良性, 意義不明, おおよそ病的あるいは 病的.変異を検出する.病的変異 は遺伝子内の欠失/重複,ミスセンス, ナンセンス, やスプライス部位を含める;エクソンや前遺伝子の欠失/重複は検出されない.
- 重症(周産期型・乳児型) 低ホスファターゼ症では,罹患者の祖先がヨーロッパ系
では,約95%に両方のアレルにALPL 病的変異が同定される. 他の病型では, 遺伝形式に応じて片方あるいは両方のアレルに ALPL 病的変異が同定される. - 片方のアレルに対立遺伝子変異が疾患を引き起こすのに充分と考えられる軽度な病型では,病的変異検出率の推定はより難しい.全体的にみて, ~50% で両方のアレルに ALPL 病的変異 (複合へテロ接合体 あるいはホモ接合体)を有し; 40%-45% では片方のアレルに病的変異のみが同定される.軽症であるほど 高い割合で片方のアレルにALPL 病的変異 が同定される.
- 標的遺伝子欠失/重複検査は 遺伝子内の 欠失 あるいは重複を検出する. 1つのエクソンの欠失 あるいは重複は,1エクソンの欠失や重複を検出するために定量PCR, ロングPCR, MLPA (multiplex ligation-dependent probe amplification), や遺伝子特異的なマイクロアレイが活用される.
- 標的遺伝子欠失/重複検査の検出率はあきらかではない. 欠失は数件報告されている[Spentchian et al 2006, Mornet 2015] (参照 www?.sesep.uvsq.fr) .
感度と特異度を含んだ検査の特質に関する情報は以下.参照 Clinical Utility Gene Card [Mornet et al 2014] .
臨床像
低ホスファターゼ症の臨床症状は,妊娠中より骨の石灰化がみられる死産の方から,成人後期に下腿の病的骨折の方まで及び幅が広い.[Whyte 1994].
低ホスファターゼ症の典型例. さまざまな重症度や各年齢でのくる病あるいは骨軟化の臨床像を有する.家系内においても, ヘテロ接合あるいはホモ接合により家系内構成員は複数の病型を有するかもしれない.骨石灰化がない死産が最も重症な表現型である.血清ALPが上昇しない逆説的なくる病(栄養性あるいは 腎性くる病)像が典型的である.年長の成人に生じる大腿骨頭,脛骨や中足骨といった下腿病的疲労骨折が軽症である.
すべての罹患者で
- 骨や歯の石灰化が.欠如している.
- 血清 アルカリホスファターゼ (ALP) 活性:低下.
組織学的評価.
- 骨の組織学的所見では,成長板でくる病病変を認める.破骨細胞の組織化学的検査では,細胞膜関連ALP活性の欠損が明らかになる.破骨細胞と骨芽細胞は正常に見える.
- 歯の組織学的所見では,セメント質の低下は明らかであり,疾病の重症度に応じ変化する.
病型による特定の表現型は,以下の通りである
- 周産期型(重症型)低ホスファターゼ症の典型的な例は,出生前超音波検査により同定される.妊娠は死産となることもある.小さい胸腔と短くて曲がった肢は出生児,死産児の両方で認める.胸郭動揺(フレイルチェスト) があるかもしれない.(図 1A).周産期型低ホスファターゼ症の乳児は肺機能不全が最も多い死因となる.高カルシウム血症の頻度は高く,無呼吸またはけいれんに関連する.
- 周産期型(良性)低ホスファターゼ症 は,通常出生前超音波検査により同定される. 長管骨の短縮や屈曲はあるが石灰化は正常あるいは軽度減少する.出生後,重症型低ホスファターゼ症 に見られる臨床像は減少し骨病変は徐々に改善するPauli et al 1999, Wenkert et al 2011]
- 乳児型低ホスファターゼ症 は,出生時は正常のことがある.臨床症状は生まれてから6ヶ月の間でくる病に類似する症状で認められる(図 1A).低石灰化骨はレントゲン像とともに大泉門の開大や縫合の拡大がある. 頭蓋骨縫合早期癒合および頭蓋内圧亢進は予測される合併症である.
臨床的重症度は,肺機能不全の程度に依存する.乳児型の死亡率は高く, 50%の患児は肋骨の石灰化 ができないため呼吸不全を来す.他の合併症として高カルシウム血症,易刺激性, 補乳力低下, 成長障害, 筋緊張低下, まれにビタミンB6-依存性けいれんを来す (参照 臨床的マネージメント) .年長者では腎障害を伴う.
- 小児型低ホスファターゼ症 は,年齢より骨密度が低いことに由来する原因不明の骨折からくる病まで及び臨床像が広い.罹患児は,通常切歯で始まる乳歯の早期脱落(5歳までに)が特徴的で,脱落歯には歯根が残っている.より重症度が高い罹患児は低身長で歩きはじめが遅く,筋障害によるアヒル歩行に進展する.骨の痛みや関節痛は典型的である.骨幹および骨幹端骨折が起こることがある.
- 成人型低ホスファターゼ症は,ときに既往として小児期に一時的に生じた「くる病」および/あるいは乳歯の早期脱落と関連する.永久歯の早期脱落もよくみられる. エナメル質形成不全や歯の動揺の報告はあるが,青年期および成人期において他の歯科的合併症は少ない.多くは,中年期において下肢の疲労骨折と偽骨折で認められる.足の痛みや,中足骨での疲労骨折が多い.ももおよび股関節の痛みは大腿骨骨幹部の側皮質での偽骨折("Looser zones") を反映することがある (図 1C). 軟骨石灰化症および骨関節症は年齢と共に進行する.骨軟化症は,成人型低ホスファターゼ症と歯限局型低ホスファターゼ症とを区別する.
- 歯限局型低ホスファターゼ症 は, 低ホスファターゼ症の上記の病型においても認められる歯の症状を単独所見として認め,他の骨格系の異常はない.乳歯の早期脱落および/または重症虫歯がみられ,最も頻繁に切歯が失われる.
遺伝子型と臨床型の関連
多くの低ホスファターゼ症罹患者「個別」の遺伝型を有し, 遺伝型-表現型の関連性の評価は難しい.しかし,部位特異的変異誘発研究により変異アレルが産生する明らかな残存酵素活性やドミナント・ネガティブ効果(参照 分子遺伝学)が示される.重症型表現型は優性型低ホスファターゼ症にあるドミナント・ネガティブ効果よりは,劣性型低ホスファターゼ症にある残存酵素活性と関連する. [Fauvert et al 2009]. 臨床像は残存酵素活性だけでなく変異部位が報告される.いくつかの変異はwww.sesep.uvsq.fr.で確認できる.
命名法
低ホスファターゼ症という名称は,血清リン濃度を反映しているというよりは,ホスファターゼ活性が低いことに由来する.
低ホスファターゼ症は,遺伝素因による分類では,代謝性骨疾患,骨系統疾患,骨幹端異形成症,歯の疾患または細胞外マトリックス中の膜結合型の細胞外酵素活性に関わる疾患として位置づけられている.
有病率
カナダのオンタリオでの小児病院病歴記録に基づくと,(常染色体劣性)周産期型,乳児型低ホスファターゼ症の出生率は10万人に1人と推定された[Fraser 1957].この推定値にHardy-Weinberg式を適用すると,病的なALPL変異のヘテロ接合体頻度はカナダのオンタリオでは約150人に1人である.
カナダのメノナイト(メノー派)では,低ホスファターゼ症(周産期重症型)の患者は2500人に1人で,保因者頻度は25人に1人となる.
フランスやヨーロッパでの分子遺伝学的検査により, 重症の病型の有病率は30万人に1人と推定される.[Mornet et al 2011].軽症の病型 (周産期良性型, 小児型, 成人型ならびに 歯限局型低ホスファターゼ症)の罹患率は,ヘテロ接合体が低い選択圧を伴い発症しているので6300人に1人よりも高いと予測される[Mornet et al 2011].この重症病型の有病率推定値にHardy-Weinberg式を適用すると,フランスでのALPL病的変異のヘテロ接合体の頻度は275人に1人である.
日本では, c.1559delT病的変異について,ホモ接合の頻度が900,000人に1人[Watanabe et al 2011])であることと,日本人患者においてアレル全体のうち,c.1559delTの割合は 40.9% [Michigami et al 2005])であることを基に,重症型 低ホスファターゼ症の出生率は,150,000人に1人と推定される.
中国では いくつかの病的変異が報告されている [Wei et al 2010, Zhang et al 2012, Yang et al 2013] .しかし出生率は不明である.
アフリカでは, 北アフリカ以外の報告はない. しかし,確定診断のバイアスも有している.アフリカ系アメリカ人の低ホスファターゼ症症例はまれである.この母集団での病的変異は欧州での混合として表されると想定される.
遺伝学的関連疾患
ALPLは低ホスファターゼ症以外に関連がある表現型のある疾患はGeneReviews にはない.
鑑別診断
低ホスファターゼ症の鑑別診断は,診断される年齢によって異なる.低ホスファターゼ症と他の疾患を鑑別する臨床的特徴には,出生前とともに出生後直ちにみられる骨石灰化減少,出生後にみられる血清カルシウムとリン濃度の上昇,加えてもちろん持続する血清ALP活性の低値が挙げられる.
出生前(子宮内).早期の出生前超音波検査は,低ホスファターゼ症とともに,骨石灰化の欠損を来す骨形成不全症(OI)II型,タナトフォリック骨異形成症 (thanatophoric dysplasia),屈曲肢異形性症 (campomerlic dysplasia),軟骨異形成症 (chondrodysplasias) が考慮される.超音波の読影に長けた人はこれら疾患の識別に困難を要しない.胎児期のX線で認識される周産期型低ホスファターゼ症に典型的な骨の骨塩低下は,他の疾患との鑑別診断に役立つ.
出生時.外見上は鑑別が難しいが, X線像からOI II型, タナトフォリック骨異形成症, 屈曲肢異形性症や軟骨異形成症と低ホスファターゼ症 とは容易に鑑別が可能である.診断が疑わしい場合,血清ALP活性や専門の生化学検査(血清PLP濃度,尿中PEA濃度) の測定は,分子遺伝学的検査の確定診断中に,本症の診断を示唆する.
乳児期・小児期.易刺激性,栄養不良,発育不全,筋緊張低下およびけいれんは,乳児型低ホスファターゼ症とともに先天性代謝異常,有機酸血症,一次性および二次的くる病,虐待および非偶発外傷など幅広い鑑別診断が入ってくる.成長障害,原因不明のけいれん,事故でない骨格の損傷では,血清ALP活性を検査項目に入れ的確な小児正常基準値を用いることで,血清ALP活性低値により,乳児型低ホスファターゼ症は疑われる.
- 難治性けいれんが,早期の低ホスファターゼ症において,理学的・画像所見よりくる病所見が明らかになる前に生じているかもしれない.
- くる病は,理学的や画像的所見で診断される早期の低ホスファターゼ症の病像である.しかしながら,原因が栄養かつ/あるいはビタミンD欠乏,ビタミンD抵抗や,腎性骨ジストロフィーによるくる病と,低ホスファターゼ症とは検査所見により容易に識別される.くる病では以下の検査所見が特徴である:
- 血清アルカリホスファターゼ活性高値,
- 血清カルシウムとリン濃度低値,
- 血清ビタミンD濃度低値,
- 血清副甲状腺ホルモン濃度高値.
- 血清アルカリホスファターゼ活性高値,
- 骨形成不全(OI)(典型的には乳児期のIII型あるいはその後のIV型)は,低ホスファターゼ症と臨床的に似ている.
・象牙質形成不全症Dentinogenesis imperfecta (DI)は,OIの一部あるいは個別の所見としてみられるが,低ホスファターゼ症の歯科所見から鑑別可能である. - 鎖骨頭蓋異形成症Cleidocranial dysostosis は,泉門および頭蓋縫合の閉鎖遅延,鎖骨欠損,恥骨枝での骨化不全,乳歯および永久歯の萌出遅延を特徴とする.骨格形成異常は,臨床検査と骨の検討により低ホスファターゼ症と鑑別可能である.
- 歯形成不全は早期の歯脱落はなく,エナメル質の低形成は歯限局型低ホスファターゼ症と容易に鑑別できる.
- Stuve-Wiedemann症候群 (OMIM 601559) (シュワルツ・ヤンペル(Schwartz-Jampel)症候群)は,体温調節不全, 反射減弱,筋緊張亢進を有しているが, 周産期重症型低ホスファターゼ症と呼吸不全, 長幹骨の彎曲, 骨幹端異形成, 年齢に比した骨密度の低下, 易骨折性.の症状は共有している.
- Cole-Carpenter症候群 (OMIM 112240) は,骨変形,多発性骨折,眼球突出, 浅い 眼窩, 眼窩縫合早期癒合, 前頭隆起および水頭症を特徴とする.
- Hadju-Cheney症候群(OMIM 102500) は,成長障害,特徴的な顔貌,初期の歯喪失,泌尿生殖器の奇形,骨粗鬆症,病的骨折,Wormian骨格,縫合線での骨化不全,頭蓋底陥入症,脊椎の異常,関節弛緩症,曲がった骨,短い遠位側指,先端骨溶解症および多毛を特徴とする.
- 特発性若年性骨粗鬆症(IJO) (OMIM 259750)は,典型的には思春期直前に骨折と骨粗鬆症でみられる.通常,骨折易罹患および骨粗鬆症は思春期に自然に治る.病因は不明である.
- 腎性骨ジストロフィは,腎障害を伴う晩期の小児型と混同される可能性がある. しかし,特徴的血液生化学所見により鑑別が可能である.
- 非偶発外傷(児童虐待).骨形成不全症のように,既往歴,家族歴,診察所見,ルーチン検査所見,画像診断および臨床経過はすべて,低ホスファターゼ症と児童虐待との鑑別に寄与する.多発骨折は低ホスファターゼ症の中でそれほど典型的ではない.家族歴では,周産期型(重症)が常染色体劣性の疾患であるという点で特に有益かもしれない. また,小児型,成人型および歯限局型は常染色体優性遺伝疾患である.説明できない小児期の骨折が契機で,低ホスファターゼ症と診断された報告は1家系のみである. [Lia-Baldini et al 2001]. この状況では連続した血清ALP活性の測定で低ホスファターゼ症と鑑別するのに充分である.
- 偽性低ホスファターゼ症は,臨床像,生化学およびX線所見において乳児型低ホスファターゼ症に類似し,血清ALP活性は正常な範囲である点が唯一異なる.
成人期と歯限局型低ホスファターゼ症
- 変形性関節症および偽痛風(ピロリン酸カルシウムへの結晶が沈着して起こる)は,成人型低ホスファターゼ症でも認め,病歴および検査所見からより一般的な疾患と鑑別される.
- 骨減少症/骨粗鬆症は,成人型低ホスファターゼ症と区別される必要がある. この点で,ビスフォスフォネートは禁忌とされる( (参照 臨床的マネジメント, 回避する薬剤/環境).
- 歯周病は重度の歯肉炎による歯槽骨喪失を認める点で低ホスファターゼ症と鑑別することは難しいかもしれない.しかし,歯肉炎症は歯限局型低ホスファターゼ症にまれである.家族性歯周病は,常染色体優性形式 (OMIM 170650)をとり,あるいはエーラス・ダンロス症候群血管型,歯根型[OMIM 130080]のような結合組織異常症の症状の一部として,またはELA2関連性好中球減少症のような好中球減少症のような常染色体優性形式 (OMIM 311750)を示す.エーラス・ダンロス症候群VIII 型では正常の歯根も含めた歯欠損が見られるかもしれないが,血清ALP活性の低値の有無で区別する.
早期に歯脱落および歯周病の原因となる稀な常染色体劣性遺伝性疾患はPapillon-Lefevre症候群 (OMIM 245000) やジペプジルペプチダーゼ1をコードするCTSC遺伝子の病的変異により発症するHaim-Munk症候群(HMS) (OMIM 245010).歯周病は歯限局型低ホスファターゼ症よりも早期に発症し,重症となる.Papillon-Lefevre 症候群と HMS通常それぞれ手掌角化症と関連付けられる点で,歯限局型低ホスファターゼ症と鑑別できる.どちらかの疾患が考慮される時,血清ALP活性の測定は理にかなっている.
- 象牙質形成不全Dentinogenesis imperfecta (DI)は,骨形成不全症に関連の症状かどうか,あるいはDSPP (OMIM 125420) 病的変異による[Rajpar et al 2002].血液生化学所見により,歯限局型低ホスファターゼ症と容易に鑑別可能である.
臨床的マネジメント
最初の診断時における評価
低ホスファターゼ症と診断された個々人において疾患の程度を明らかにするために,以下の評価の実施が推奨される.:
- 腎機能を評価する血中尿素窒素および血清クレアチニン濃度
- 血清カルシウム,リン,マグネシウム濃度
- 血清中25(OH)および1,25(OH)2ビタミンD,nPTH(副甲状腺ホルモン, N末端)によるくる病の評価
- 肺機能の評価を行い,周産期型の予後,周産期重症型・周産期良性型を区別する 頭部X線により幼児型低ホスファターゼ症の児の頭蓋骨癒合を評価する
- 歯科評価
- 整形外科評価
- 臨床遺伝専門医 および/あるいは 遺伝カウンセラーの受診
症状に合わせた治療
すべての年齢において,疾患に関連する合併症を最小化するための補助療法に注目している.
さまざまな年齢における複数領域の専門的な評価が含まれる:
- 内分泌では,骨ホメオスタシス状態を最大限に活用し,悪化する治療を避ける
- 腎臓では,カルシウムホメオスタシスを評価し,腎石灰化症を検査する
- 神経ではけいれんの予防的予測的治療や筋萎縮を管理する
- 脳神経外科あるいは頭蓋・顔面チームにより,偽頭蓋骨融合症を管理する
- 整形外科では原発性あるいは二次性の骨病変を管理する
- 物療医学やリハビリテーション (PM&R) では, 自主的な可動性を十分に活用できるように理学療法や作業療法が行われる
- 疼痛管理
- 心理的サポート
- 小児ならびに成人歯科による歯脱落の管理
複雑で相互に関係のある医療事項への治療には,複数領域の専門家が関与し個々の管理や社会資源によるサポートに関わる.
酵素補充療法.
アスホターゼ アルファ (ストレンジック™)による組織非特異的アルカリホスファターゼ (TNSALP) 酵素補充療法 (ERT)の出現は,周産期重症型や乳児型 HPP患児の自然歴を変えつつある.長期効果はまだ不明な点もあるが,もはや薬剤が開発された今,新しい表現型となる「治療後の周産期型と乳児型 HPP」により,以前使用された病型である 周産期致死型HPPは一般的にあてはまらない.
2015年10月には, FDA はアスホターゼ アルファを 周産期型, 乳児型や若年発症のHPPへの治療薬として承認した[Alexion –10-23-2015].(訳注 日本においては,アスホターゼ アルファ(ストレンジック™)は世界に先駆けて2015年7月に承認された)
- 周産期型/乳児型 HPP 研究成果 .2つの前向き単一群研究 (生存率分析を用いたヒストリカルコントロールと比較),周産期重症型/乳児型HPP 68 名(治療開始年齢: 1 日– 78 ヶ月)少なくとも 24週にわたる TNSALP ERT が完了した(≤9 mg/kg 毎週, 皮下注射) [Whyte et al 2016] (最終結果).
- 生存率.呼吸的補助が必要とした群で(n = 26名), 21名 (81%) が評価最終まで生存 (平均 3.2 年), ヒストリカルコントロールと比べると1:20 (5%) の生存率であった.
- 周産期型/乳児型 HPP 68名にアスホターゼ アルファ ERTを開始した混合コホートでは,人工呼吸器を必要とした54名中,最終確認で91% が生存し,85% が呼吸器不要となっていた.48名のヒストリカルコントロールでは 27% が生存し ,25% が呼吸器不要となっていた[Whyte et al 2016] (最終結果) .ERT の臨床研究では発達の目安や肺機能の改善がみられていた[Whyte et al 2012].
- 骨病変.上記研究の64名 の患児と3番目の若年性 HPPに向けた前向き非盲検研究における4名(計68名) に対して,骨の画像評価を 7 段階で評価する Radiographic Global. Impression of Change(RGI-C)スケールを用いてHPP関連くる病を評価した. 画像上の変化が少なくとも +2 の患児("効果有り"と定義) は,最終で治療した50名/68 名(74%) に認めた (参照 図 2), (対照すべきヒストリカルコントロールはない).周産期型/乳児型HPP18名 が治療中に骨折を経験しており,アスホターゼ アルファの骨折への効果はまだ明らかではない[Whyte et al 2016] (最終結果).
- 若年発症HPP 研究成果. 若年発症HPP8名と周産期/乳児期発症HPP5名を含む前向き非盲検, 単一群研究では,治療開始年齢は6-12 歳である.若年発症HPP患児 では,TNSALP ERT (6 mg/kg 毎週, 皮下注射)を少なくとも48ヶ月を終了した.若年発症HPP患児 8名を32名のヒストリカルコントロールと比較した. 画像による RGI-Cスケールでは, 患児 8名すべてで効果有りと考えられた.ヒストリカルコントロールのうち2名 (6%)は 54ヶ月の段階で+2 以上の改善を認めている. 歩行スコアはmodified Performance-Oriented Mobility Assessment(mPOMA-G)と 6分間歩行試験 (6MWT)で評価し,アスホターゼ アルファ治療患児で歩幅は改善していた. 6MWTも患児6名中6名で 48ヶ月で正常域に改善していた.このデータは若年発症HPPにおける骨折へのアスホターゼ アルファの効果を評価するためには現在のところ不十分である [Whyte et al 2016] (最終結果) .
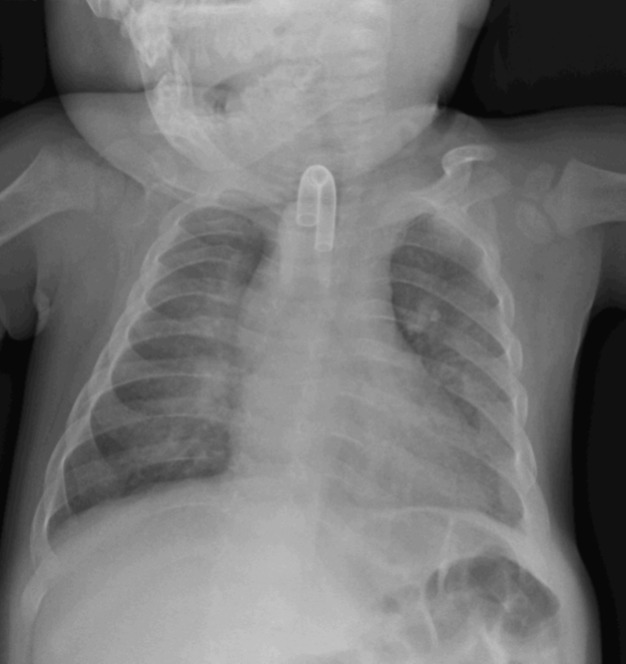
図 2.低ホスファターゼ症治療後の レントゲン像
A. 患児 (図 1A)のアスホターゼ アルファ 酵素補充療法12月後
B. 気管切開チューブが疾患治療後の病像である喉頭軟化症や気管支軟化症に対し置かれている
C. くる病肋骨や骨幹病変は改善した
D. 2つの骨端成長領域(上腕骨近位) は正常
酵素補充療法が利用できない, あるいは使用典型例でない場合:
周産期型. 周産期型 HPPで新生児期すぐにアスホターゼ アルファ ERT治療を実施した経験は限られており,そしてこの治療は容易に利用できない.複数領域の専門的評価により新生児期すぐに周産期重症型HPPと診断されると,患児や家族への緩和ケアや支持的管理が選択肢として残っている.
乳児型. 乳児例では致死率は高く,死亡の50%は肋骨の骨塩低下によって引き起こされる呼吸不全により死亡している.ERTができないときには支持的管理がある.
- カルシウムホメオスタシス.手に負えない高カルシウム血症/高カルシウム尿症により管理がさらに複雑化し, 最適な管理戦略は明らかではない.高カルシウム血症/高カルシウム尿症は典型的には水分補給やフロセミド治療に抵抗性であり,ビスフォスフォネート は禁忌である (参照 回避するべき薬剤/状況:). ERTができないときに, カルシトニンやステロイド は限られた効能ではあるが短期間に試みられている.[Deeb et al 2000].
- けいれん.現在のところ,けいれん発作はビタミンB6(ピリドキシン)治療に反応する可能性がある.ピリドキサールリン酸(PLP)(アルカリホスファターゼの天然基質のうちの1つ)は,ピリドキシンが本質的な酵素活性を媒介する活性化合物である.中枢神経系でのPLP欠乏は,神経伝達物質(GABA)の合成を抑制し,発作閾値を下げる可能性がある.
- 乳児型の頭蓋骨癒合症には幅がある. 同定された際には脳神経外科医による管理は慎重に行う.頭蓋骨癒合症による二次的な脳圧亢進は外科的減圧の適応である.
- 歯科検診は,乳歯の保護(栄養管理を含めて)永久歯列の保護や置換.を行うために1歳から開始するのが重要である.
小児型 および成人型 低ホスファターゼ症
- 骨関節炎は,NSAIDs が反応するかもしれない.
- 骨痛および骨軟化症は,対症療法で管理される.非ステロイド性抗炎症薬(NSAIDs)は有用である.Girschick et al 2006]. 低ホスファターゼ症でのビスフォスフォネートによる治療は相対的禁忌となる.(参照 回避する薬剤/環境).
- 偽骨折および疲労骨折は管理が難しい.内固定は最適な整形外科的管理として指摘されている.足の矯正器具は,成人の場合足根骨骨折および偽骨折の管理に役立つ可能性がある.
一次症状への予防
負担が掛からない身体活動や運動は全体的な骨の健康を改善するかもしれない.低ホスファターゼ症を周知する専門医師による管理が勧められる.
二次症状への予防
サプリメントによるカルシウム添加やビタミンDを用いた治療は,成人での二次性副甲状腺機能亢進症を予防できるかもしれない. 低ホスファターゼ症を周知する専門医師により詳細な追跡評価をすべきである.
管理(サーベイランス)
低ホスファターゼ症小児において,小児歯科の診察は1歳に開始され,年に2度行われるべきである.
乳児型低ホスファターゼ症の小児では,頭蓋骨癒合症による頭蓋内圧亢進を来す高いリスクがあり,この合併症のためモニターされるべきである.
回避する薬剤/環境
ビスフォスフォネートは,低ホスファターゼ症には相対的禁忌である. ビスフォスフォネート製剤による副反応は重症の乳児型の小児や低ホスファターゼ症および骨軟化症の成人では同定されていないが[Deeb et al 2000],ビスフォスフォネートの構造に基づく理論から懸念されている. ビスフォスフォネート製剤でのリン酸モチーフはTNSALPの天然基質である無機ピロリン酸(PPi)と同様の構造を有する.それ故, ビスフォスフォネート投与は火に油を注ぐことと同様である.低ホスファターゼ症や骨軟化症でビスフォスフォネートを治療された成人で, 側面での大腿骨転子部偽骨折が報告されている [Whyte 2009].成人型低ホスファターゼ症の罹患率は不明であるが,診断されていない成人患者の多くは間違いなくビスフォスフォネートで治療され,この予期せぬ併発症の頻度は不明である.
過剰量のビタミンD は,高カルシウム血症を伴った乳児型低ホスファターゼ症において高カルシウム尿症血症/尿症を悪化させる.
テリパラチド (副甲状腺ホルモンの N 末端から 34 番目までのアミノ酸で構成される組換えタンパク) の高用量投与でラットでは骨肉腫が発症し,さらに放射線に起因する骨肉腫(小児における成長板部位の腫瘍)のリスクを上昇するかもしれない.この薬剤は 低ホスファターゼ症.の小児には禁忌である.
リスクのある親族への検査
リスクのある親族への検査に向けた遺伝カウンセリングについては,遺伝カウンセリングの項を参照のこと
研究中の治療
酵素補充療法 (ERT). 常染色体劣性低ホスファターゼ症 はとてもまれで,重症である.周産期型ならびに
乳児型 HPPへのアスホターゼ アルファによるERTの長期治療効果はまだ明らかではない.IV 相臨床試験はFDA 承認までに最大78ヶ月まで治療されている.
小児,思春期,成人の臨床研究は進行中であるため,軽度の常染色体劣性や常染色体優性小児型ならびに成人発症 HPPでは, アスホターゼ アルファに関する情報は限られている.
骨髄移植(造血幹細胞移植) 8か月の重症低ホスファターゼ症の女児に行われ,臨床的にも放射線的にも長期にわたる顕著な効果を得られた. [Whyte et al 2003].移植の7年後に患者は元気で成長を続けており,軽症の小児型ホスファターゼ症の臨床病型を有していたと報告された [Cahill et al 2007] .他の研究では 8ヶ月患児に骨髄移植と同種間の間葉系幹細胞移植を行ったところ呼吸機能の改善が見られた [Tadokoro et al 2009] .しかしながらこの患児は治療関連白血病を発症している [Taketani et al 2013].すでに骨髄移植を受けている患者への増幅したex vivo間葉系幹細胞移植は,骨ミネラル化,筋肉密度,呼吸機能,知的発達や寿命を改選した. [Taketani et al 2015] .
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
周産期型および多くの乳児型低ホスファターゼ症は,常染色体劣性形式を示す.
軽症の病型,特に成人型および歯限局型ではALPL変異がTNSALP活性に及ぼす影響により常染色体劣性または常染色体優性形式が決まる[Fauvert et al 2009].
患者家族へのリスクー常染色体劣性遺伝形式
発端者の両親
- 多くの場合,罹患者の親は潜在するヘテロ接合体である(すなわち,片方のアレルに変異を有する保因者)である.
- とてもまれではあるが,患児の片親のみがヘテロでALPL 病的変異.を有している.
- Taillandier et al [2005] とZhang et al [2012] は,それぞれ常染色体劣性 低ホスファターゼ症患者の2番目の病的変異がde novo で生じる症例報告をしている.
- 1番染色体の片親性ダイソミーが劣性低ホスファターゼ症により発症した2例が報告されている[Watanabe et al 2014, Hancarova et al 2015].
- ALPL 病的変異部位により,ヘテロ接合体(保因者;キャリア)である両親は,単に生化学所見を有する無症候性あるいは患児よりも軽い症状を呈する.軽症の両親を有する家系において遺伝形式を決定することは難しく,ときに常染色体優性遺伝を考慮する.
発端者の同胞
- 妊娠した時点で,罹患者の兄妹はそれぞれ,25%は罹患者,50%はヘテロ接合体保因者,25%は保因者でない非罹患者になる.
- ヘテロ接合体(保因者)は変異部位により,生化学(検査所見)的に異常はあるが,臨床的に異常を認めない無症候性の場合と,あるいは発端者より軽い症状を呈する場合がある.
発端者の子孫.
常染色体劣性 低ホスファターゼ症を伴う方の子孫は,ALPL遺伝子の病的変異の絶対保因者(キャリア) である.
発端者の他の家族
発端者の親のそれぞれの兄弟は, 50%で病的変異保因者のリスクを有する.
保因者(ヘテロ)検出
罹患する可能性のある家系内での保因者診断は,家系内でのALPL 病的変異同定が必要である.患者家族のリスクー常染色体優性遺伝
発端者の両親
- 低ホスファターゼ症の常染色体優性遺伝形式と解釈された個人の大半は,症状があるかもしれない片親からの変異を受け継いでいる.
- 常染色体優性遺伝形式を呈する低ホスファターゼ症では,新規(de novo)変異は報告がない.
- 発端者に見られた遺伝子変異をどちらの両親からの白血球由来のDNAに検出できない場合,片親の生殖系列モザイクまたは発端者での新生(de novo)変異という2つの可能性がある.
- 発端者の両親の評価への推奨 低ホスファターゼ症の兆候をみつける病歴および検査評価の注意深い検討を含んでいる.両親の評価により,片親が罹患していることが判明するかもしれない.しかし,医療従事者がこの疾患を鑑別に挙げない場合,低い浸透率や症状が軽いため診断されていない場合がある.したがって,適正な評価が行なわれるまでは家族歴がないとは正確には言えない.
発端者の同胞
- 発端者の同胞のリスクは,発端者の両親の遺伝的状態によって異なる.
- 発端者の親がALPL 病的変異を有する場合,同胞が病的変異を有するリスクは50%である.
臨床的重症度は,罹患家系のメンバーでしばしば類似するが,低下している浸透率と発現の多様性から,家族歴や分子遺伝学的検査からも確実な予測はできない.
発端者の子孫.
常染色体優性遺伝形式となる低ホスファターゼ症の患者さんの子どもは,それぞれ50%の割合で遺伝子変異を引き継ぐ.
他の家系構成員.
他の家系構成員へのリスクは,発端者の両親の状況によって変わる.親がALPL 病的変異を有すると分かると,対象家族はリスクを負う.
遺伝カウンセリングに関連した問題
遺伝形式の決定 は,家系により,罹患児の両親の症状があっても軽度であり症状の出現に幅が大きく難しい.
de novo 病的変異が考慮される家系 常染色体優性病型を来す発端者の両親が病的変異を有さず症状がない場合 , この病的変異はほぼde novoである しかし, 異なる父性,母性(すなわち生殖補助医療による)あるいは告知されていない養子といった非医療的理由によってもあり得る.
家族計画
- 遺伝的リスクの評価,保因者診断,および出生前診断が実施できるかの検討は妊娠前が望ましい.
- 罹患もしくは保因者の可能性がある若年成人に対しては,子どもへの遺伝の可能性や挙児に関する選択肢を含めて遺伝カウンセリングの提供がなされるのが適切である.
DNAバンク は主に白血球から調製したDNAを将来の使用のために保存しておくものである.検査法や遺伝子,変異あるいは疾患に対するわれわれの理解が将来進歩するので,DNAの保存は考慮に値する.
出生前診断
事前リスクが高い妊娠(家族歴に基づいたリスクが上昇すると想定された妊娠)
- 分子遺伝学的解析.家系内の罹患児ですでに遺伝子変異が同定されて可能となる.
- 胎児超音波検査.周産期型低ホスファターゼ症の再発を出生前超音波検査により確認できる可能性がある.骨塩低下,小さい胸腔,短い長幹骨および弯曲は,常染色体劣性・重症型低ホスファターゼ症の典型的像である.長幹骨弯曲は,小児型または成人型低ホスファターゼ症の患者の罹患した兄弟や児においても出生前に報告されている.しかし,妊娠後期から出生後に症状が改善する非致死性低ホスファターゼ症となる周産期型良性低ホスファターゼ症である病型があり,長幹骨弯曲から周産期型低ホスファターゼ症と診断はしない[Moore et al 1999, Pauli et al 1999, Wenkert et al 2011].
- 生化学検査.羊水,羊膜細胞および絨毛検体中のALPの濃度は,誤読(特に非罹患ヘテロ接合体と区別する際に)する可能性がある.出生前診断での確認手法として分子遺伝学的検査が望ましい理由である[Mornet et al 1999].
事前リスクが低い妊娠(リスクが想定されない妊娠)
- 胎児超音波検査. 周産期型低ホスファターゼ症は,出生前超音波検査により他の骨系統疾患と鑑別するが,屈曲した長幹骨への対応が考慮されるべきである.骨塩低下,小さい胸腔,短い長幹骨および弯曲は,常染色体劣性・重症型低ホスファターゼ症の典型的像である.超音波所見単独では予後の予測は難しい.
出生前超音波検査で弯曲し短縮した長幹骨は,周産期型良性,小児型および成人型低ホスファターゼ症と最終的に診断された患者においても観察されている.弯曲は出生後に改善する.ALPL分子遺伝学的検査を行うと,50% に片方のアレルに病的変異が同定され,良性表現型の確認と周産期型 (重症) 低ホスファターゼ症を除外する.
分子遺伝学
下記の記述は最新の情報が含まれているため、GeneReviewsに記載されているほかの情報と異なる場合がある
表A.
低ホスファターゼ症: 遺伝子とデータベース
| 遺伝子 | 染色体座位 | タンパク | Locus Specific | HGMD |
|---|---|---|---|---|
| ALPL | 1p36?.12 | アルカリホスファターゼ,組織非特異的アイソザイム(TNSALP) | 組織非特異的アルカリホスファターゼ遺伝子変異データベース ALPL データベース |
ALPL |
上記データは以下の標準文献である遺伝子はHGNC;から染色体座位, 座位名, OMIMから; タンパクは UniProtからより編集した.
表 B.
OMIM に掲載されている 低ホスファターゼ症
| 146300 | 成人型 低ホスファターゼ症 |
| 171760 | アルカリホスファターゼ, LIVER; ALPL |
| 241500 | 乳児型 低ホスファターゼ症 |
| 241510 | 小児型 低ホスファターゼ症 |
遺伝子構造. この遺伝子は12のエクソンから構成される.11 のコード領域のエクソンと1つの 非翻訳領域のエクソンからなる. さらなる詳細な遺伝子とタンパクの情報は 参照 表 A, .
良性対立遺伝子型 3つの良性対立遺伝子多型は,c.455G >A(p.Arg135His),c.787T >C(p.Tyr246His)およびc.876A >G(p.Val505Ala)である.多くのエクソンやイントロンでみられる塩基変化は,ALPL 遺伝子変異データベースに多型として報告されている.
疾患関連性対立遺伝型 これまで,300 を超えるALPL遺伝子変異報告され北アメリカ, 日本, ヨーロッパ人が多い.最遺伝子変異リストは,オンラインで取得可能である 参照 www.sesep.uvsq.fr. (参照 表 A.)
病的な遺伝子変異はALPL遺伝子の12のエキソンの全体にわたっている..ミスセンス変異は 変異全体の74.6%,残り小欠失・挿入(13.3%), スプライシング部位変異(6.0%),ナンセンス変異(3.7%),大きい欠失(1.3%),および主たる転写開始部位に影響する塩基置換である.これらの遺伝子変異の多様性は,極めて変化に富む臨床症状や数多くの複合ヘテロ接合性遺伝子型をもたらしている.
表4.
代表的な ALPL 変異部位
| Variant Classification | DNA (塩基置換) | タンパク質(アミノ酸置換) | Reference Sequences |
|---|---|---|---|
| 良性 | c.455G>A | p.Arg152His | NM_000478?.3 NP_000469?.3 |
| c.787T>C | p.Tyr263His | ||
| c.1565T>C | p.Val522Ala | ||
| 病的 | c.571G>A | p.Glu191Lys | |
| c.979T>C | p.Phe327Leu | ||
| c.1001G>A | p.Gly334Asp | ||
| c.1133A>T | p.Asp378Val | ||
| c.1559delT | p.Leu520ArgfsTer86 |
アミノ酸残基はシグナルペプチドの最初から番号が振られる.変異の分類やこの表にある変異は著者により規定した.GeneReviews スタッフは独自に分類を確認していない.
記載法: GeneReviews はHuman Genome Variation Society (www?. hgvs.org). の標準化に対応している.
正常遺伝子産物 ALPL遺伝子は,ALP,組織非特異的アイソザイム(TNSALP), 肝,腎臓および骨に存在するアイソザイムをコ一ドする..アルカリホスファターゼ は, 17アミノ酸からなるシグナルペプチド,507アミノ酸からなる成熟ペプチドの計524アミノ酸からなり,ホモダイマー(二量体)で機能する.この酵素はPPi,PLPおよびPEAとともに天然基質で(脂質)膜結合する表面酵素として機能する.
異常遺伝子産物. 病的変異は様々な結果や時に蓄積して表現される.すなわち,酵素活性の低下あるいは欠如や2量体を形成できず, 細胞コンパートメントにとどまり,表現型を表す生理学的活性の最終目的地である細胞膜に到達しない [Cai et al 1998, Fukushi et al 1998, Shibata et al 1998, Watanabe et al 2002, Brun-Heath et al 2007, Sultana et al 2013,Numa-Kinjoh et al 2015].
遺伝型・表現型の関連は,部位特異的変異誘発や酵素の3Dモデル化で研究される.これらの研究成果は重症や中等症アレルにより産生する残存酵素活性や優性遺伝形式をとるドミナントネガティブ効果を明らかにした [Fukushi et al 1998, Shibata et al 1998, Zurutuza et al 1999, Mornet et al 2001, Watanabe et al 2002, Nasu et al 2006, Brun-Heath et al 2007, Fauvert et al 2009]. しかしながら,このような 病的変異の感度を確実に予測しない.
更新履歴
-
Gene Review著者: Etienne Mornet, PhD, Mark E Nunes, MD
日本語訳者:渡邉 淳 (日本医科大学付属病院遺伝診療科)
Gene Review 最終更新日: 2016.2.4. 日本語訳最終更新日: 2016.5.12.(in present)