ミトコンドリア異常症概説
(Mitochondrial Disorders Overview)
[同義: Mitochondrial Encephalomyopathies, Mitochondrial Myopathies, Oxidative Phosphorylation Disorders, Respiratory Chain Disorders]
Gene Reviews著者: Patrick F Chinnery, BMedSci, MBBS, PhD, FRCPath, FRCP, FMedSci
日本語訳者: 和泉 賢一 (札幌医科大学医学部遺伝医学,NGSDプロジェクト)
Gene Reviews 最終更新日: 2014.8.14 日本語訳最終更新日: 2017.12.29
原文 Mitochondrial Disorders Overview
要約
疾患の特徴
ミトコンドリア異常症はミトコンドリアの呼吸鎖の機能障害によって引き起こされる多様な疾患群である.この疾患は細胞核内DNAもしくはミトコンドリアDNA(mtDNA)における変異によって引き起こされる.レーベル(レーバー)遺伝性視神経萎縮症(LHON)を例とする一部のミトコンドリア異常症は一種類の内臓器官のみに影響を及ぼすが,多くの疾患では複数の内臓器官に影響を及ぼし,顕著な神経と筋肉症状をもたらす.ミトコンドリア異常症はすべての年齢層にわたって発症する可能性がある.変異ミトコンドリアDNAをもつ多くの患者は多彩な症状を示し,異なる臨床的な症候群(例: キアーンズ・サイヤー症候群(KSS),慢性進行性外眼筋麻痺(CPEO),乳酸アシドーシスと脳卒中様エピソードを伴うミトコンドリア脳筋症(MELAS),赤色ぼろ線維・ミクローヌスてんかん症候群(MERRF),運動失調と網膜色素変性症を伴う神経衰弱(NARP),リー症候群(LS))を起こす.しかし,臨床的にかなり多様性が存在し,多くの患者は一つのカテゴリーでは当てはまらず、疾患表現型のオーバーラップした重ね合わせによっておこる.(これは、ミトコンドリア劣性運動失調症候群(MIRAS)が多くのミトコンドリア病の原因である核のPOLG遺伝子の変異で起こることを含む).ミトコンドリア病―ミトコンドリア遺伝子または核遺伝子を含む―の共通な臨床徴候は眼瞼下垂,外眼筋麻痺,近位筋障害と運動不耐性,心筋症,感音難聴,視神経萎縮,網膜色素変性,糖尿病が挙げられる.中枢神経への症状は変動性脳症,てんかん,認知症,偏頭痛,脳卒中様発作,運動失調,痙攣が挙げられる.妊娠中期と後期における流産はよく見られるが,認識されていない.
診断・検査
一部の患者にはミトコンドリア異常症の特徴的な症状(レーバー遺伝性視神経萎縮症, 運動失調と網膜色素変性症を伴う神経衰弱,母系遺伝リー症候群)が見られ,診断は血液より得られた分子遺伝学的検査法で病的ミトコンドリアDNAを同定することによって確定できる.そのような検査をしない多くの患者は,より構造的アプローチが必要である.そのアプローチには,家族歴や血液・脳脊髄液の乳酸濃度,神経画像検査,新規の評価,ミトコンドリアDNAもしくは核遺伝子の病的変異を調べるための分子遺伝学的検査が含まれる.検討すべき発端者の分子遺伝学的検査のアプローチは,点遺伝子変異検査,多遺伝子パネル検査(多くの遺伝子の同時検査),ゲノム検査(例えば、ミトコンドリアゲノム全体のシークエンスや核遺伝子での病的変異を同定するエキソーム解析など)である.分子遺伝学的検査が勧められなかったり診断の確定できない人では,疑われるミトコンドリア病の更なる検査として,呼吸鎖機能の筋生検を含む機能検査を含む特殊な臨床検査を行うことがある.臨床的マネジメント
ミトコンドリア異常症の臨床的マネジメントは概して支持的なものである.臨床的マネジメントは初期診断と糖尿病の治療,ペースメーカー,眼瞼下垂の矯正,白内障の眼内水晶体交換,感覚難聴の蝸牛の移植などが含まれる.複合体Iおよび複合体II欠損の患者では時にリボフラビン(ビタミンB2)の経口投与が有効なことがある.ユビキノン(コエンザイムQ10)欠損の患者は経口コエンザイムQ10療法が奏功することがある.ミトコンドリア神経性胃腸管系脳筋症(MNGIE)は造血幹細胞移植が有効なことがある.遺伝カウンセリング
ミトコンドリア異常症はおそらく核DNAもしくはmtDNAの欠損によって生じる.核DNA欠損は常染色体劣性遺伝か常染色体優性遺伝形式で遺伝することがある.mtDNA欠損は母系遺伝によって遺伝する.一般的にミトコンドリアDNA欠失は突然変異によるので,家族に1人のみであり、再発するリスクはほぼ24人に1人である.ミトコンドリアDNAの点変異や重複は母性遺伝が見られる.患者の父親が疾患を引き起こす病的mtDNA変異を保持する可能性はなく,通常は母親がmtDNA変異を保持し,症状はある場合とない場合がある.男性はmtDNA変異を子に伝えない.野生型mtDNAと病的点変異mtDNAが混在している(ヘテロプラスミー)女性は,病的mtDNA遺伝子の混在の程度が様々な状態で,変異mtDNAを子に伝えることがあり,変異したmtDNAの割合によって同じ家族の兄弟間でもかなり臨床症状が多様性に富む結果となる.ミトコンドリア異常症の出産前診断とその解釈はmtDNAのヘテロプラスミーのために困難となっている.組織特異的な病的ヌクレオチドの突然変異は稀であり,再発リスクの低さと関連している.
定義
ミトコンドリア病はミトコンドリアの呼吸鎖の機能異常によって引き起こされる臨床的に様々な機能障害の集合疾患である.ミトコンドリア呼吸鎖は好気性新陳代謝に不可欠な最終共通経路で,好気性新陳代謝に依存度が高い組織と臓器は,優先的にミトコンドリア異常症に関わっている [Wallace 1999].
70以上の異なるポリペプチドがミトコンドリアの内膜と相互作用し呼吸鎖を形成する.大部分のポリペプチドのサブユニットは細胞核の遺伝子転写物から細胞質で合成されるが,13の不可欠なサブユニットは16.5kbのミトコンドリアDNAにコードされている [Larsson & Clayton 1995].
図1にヒトのミトコンドリアDNAの構成を示す.
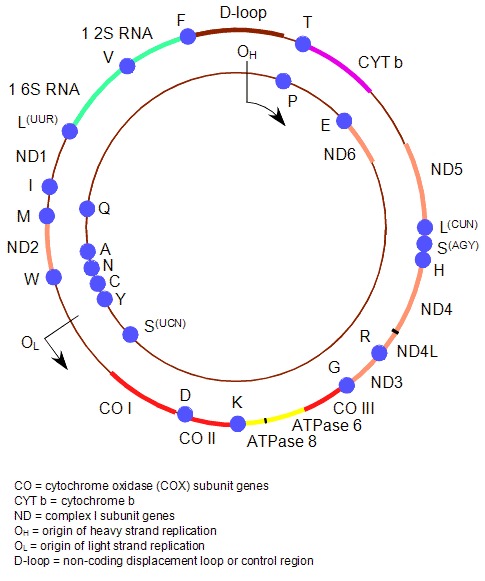
図1. ヒトミトコンドリアゲノム
- 1.1kbのDループ(遺伝子がコードされていない領域)は遺伝子転写の調整とmtDNAの複製に関与しており,この領域のみが呼吸鎖ポリペプチドの合成に直接関与していない.
- MT-ND1からMT-ND6,MT-ND4Lは複合体Iの7つのサブユニットをコードしている.
- Cyt bはmtDNAだけに コードされている複合体IIIである.
- MT-COX(cytochrome c oxidase, COX)1から3は複合体IV(チトクロームCオキシダーゼ,もしくはCOX)の三つのサブユニットをコードしている.
- MT-ATP6とMT-ATP8は複合体Vの2つのサブユニット:ATPase6, ATPase8をそれぞれコードしている.
- 二つのリボソームRNA遺伝子(12S,16S rRNAをコードしているMT-RNR1とMT-RNR2)と22のtRNA遺伝子はタンパク質をコードする遺伝子の間にコードされている.これらはミトコンドリアのタンパク質合成に不可欠なRNAである.
- OHとOLはミトコンドリアDNAの複製H鎖とL鎖の起点である.
個々のヒト細胞は出生時には通常遺伝的に同一な数千ものmtDNAのコピーを保持している(ホモプラスミー ).対照的に,mtDNAに変異を持つミトコンドリア異常症の患者は変異と正常の両方のmtDNAを個々の細胞に保持している(ヘテロプラスミー)[Holt et al 1988, Holt et al 1990].単一細胞と融合細胞を用いた研究によると,細胞が生化学的に異常なミトコンドリア呼吸鎖を示すようになるには変異mtDNAの割合がある一定値を越えなければならないことが明らかにされている(閾値効果)[Schon et al 1997].変異mtDNAの細胞内の割合は同一家族内でも個人によって異なり,また患者個人においても器官と組織で異なる[Macmillan et al 1993].これはミトコンドリア異常症を持つ患者に様々な臨床表現型が現れる一つの理由ともされる.例として,m.8993T>Gの変異を持つ患者では,変異の割合はNARP(神経性薄弱運動失調網膜色素変性症)患者よりもリー症候群患者により多く見られる[Uziel et al 1997, White et al 1999a].
他の重要なミトコンドリア機構は核遺伝子によってコントロールされている.
- mtDNA維持の障害(mtDNA欠失もしくは2次的病的mtDNA変異);
- ミトコンドリア蛋白合成障害;
- コエンザイムQ10生合成障害;
- 呼吸鎖複合体もしくはその集合体の障害.
ミトコンドリア蛋白をコードした1000以上の核遺伝子によって,分子学的診断は行われている.
疾病による2次的ミトコンドリア障害.
ミトコンドリア障害は,メチルマロニル酸尿症(ETHE遺伝子の変異による)[Tiranti et al 2009],フリードライヒ運動失調症(FXN)[Rotig et al 1997],遺伝性痙性対麻痺7(SPG7)[Casari et al 1998],ウイルソン病(ATP7B)[Lutsenko & Cooper 1998]を含む,多くの異なる遺伝子疾患によっても起き,加齢の1部分状態としても認められる.これらは厳密にはミトコンドリア病ではない.ミトコンドリア障害という言葉は通常,酸化的リン酸化に影響するミトコンドリアの1次的障害を言う。
臨床所見
ある種のミトコンドリア異常症は,眼の眼球神経障害(LHON)や,アミノグリコシド感受性もしくは非感受性の無症状難聴などのように単一の器官に影響を及ぼす.しかし多くの異常症は複数の器官に影響を及ぼし,顕著な神経障害,筋障害を現す.
ミトコンドリア異常症は幅広い年齢層にわたって現れる[Leonard & Schapira 2000a, Leonard & Schapira 2000b].最近まで、核内DNA障害は子供時代から,mtDNA障害(1次的か、核DNAの異常による2次的なもの)は子供時代後期から成人期にかけて現れると一般的に考えられていた;しかし,最近の研究で,多くのmtDNA障害は子供時代に起こり,多くの遺伝学的ミトコンドリア障害が成人期に起こることが示された.
多くの患者は独立した臨床表現型を現し,それぞれグループにまとめることができる(表1)[Dimauro & Schon 2001, Munnich & Rustin 2001].例として,キアーンズ・セイヤー症候群(KSS),慢性進行性眼筋麻痺(CPEO)[Moraes et al 1989], 乳酸アシドーシスと脳卒中様発作を伴うミトコンドリア脳筋症(MELAS)[Hirano et al 1992], 赤色ぼろ線維・ミクローヌスてんかん症候群(MERRF)[Hammans et al 1993], 運動失調と網膜色素変性症を伴う神経衰弱 (NARP)[Holt et al 1990], リー症候群 [Ciafaloni et al 1993]が挙げられる.しかし,臨床表現型は様々であり,多くの患者は適切なカテゴリーに当てはまらないことが多い.ミトコンドリア病の主要な原因として挙げられるPOLG遺伝子変異はこのことをよく示しており,おなじ遺伝子の病的変異から生じる疾患の表現型はいくつもオーバーラップしたうえで連続している(POLG関連疾患の項参照).
共通の臨床表現型は眼瞼下垂,外眼筋麻痺,近位筋障害,運動不耐性,心筋障害,感音難聴,視神経萎縮症,網膜色素変性症,そして糖尿病が含まれる.糖尿病と難聴もよく認められる臨床表現型である[van den Ouweland et al 1992].
中枢神経系に見られるのは変動性脳障害,てんかん,認知症,偏頭痛,脳卒中様発作,運動失調,痙攣である.舞踏病と認知症も顕著に見られる [Nelson et al 1995].
高い確率で起こる妊娠中期と後期の流産もまた共通に見られる特徴であるが,あまり認知されていない [Tay et al 2004].
表1 ミトコンドリア異常症の臨床表現型
| 疾患 | 主な臨床表現型 | その他の臨床表現型 |
|---|---|---|
| アルパーズ症候群 (alpers-huttenlocher syndrome; AHS) |
|
|
| 運動失調性ニューロパチー症候群(ANS):MIRAS, SCAE, SANDO, MEMSAを含む |
|
|
| 慢性進行性外部眼筋 麻痺 (CPEO) |
|
|
| カーンズ・セイヤー症候群(KSS) |
|
|
| ピアソン症候群(汎血球減少症) |
|
|
| 乳児性筋症と乳酸アシドーシス(致死性と非致死性) |
|
|
| リー症候群(LS,亜急性壊死性脳脊髄炎) |
|
|
| 運動失調と網膜色素変性症を伴う神経衰弱 |
|
|
| 乳酸アシドーシスと脳卒中様発作を伴うミトコンドリア脳筋症 (MELAS) |
|
|
| ミオクローヌスてんかん-ミオパチー-感覚性運動失調(MEMSA)1 |
|
|
| 赤色ぼろ線維とミオクローヌスてんかん症候群 (MERRF) |
|
|
| レーベル(レーバー)遺伝性視神経萎縮症 (LHON) |
|
|
CPEO = chronic progressive external ophthalmoplegia
KSS = Kearns-Sayre syndrome
LHON = Leber hereditary optic neuropathy
MELAS = mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes
MEMSA = myoclonic epilepsy myopathy sensory ataxia
MERRF = myoclonic epilepsy with ragged-red fibers
MIRAS = mitochondrial recessive ataxia syndrome
NARP = neurogenic weakness with ataxia and retinitis pigmentosa
SANDO = sensory ataxia neuropathy, dysarthria, ophthalmoplegia
SCAE = spinocerebellar ataxia with epilepsy
- MIRAS and SCAEとしても言及
鑑別疾患
乳酸アシドーシス.これらの検査値を解釈するときに乳酸アシドーシスの別の原因を除外するのは大事である.例えば,罹患した人では痙攣後,血漿と脳脊髄液(CSF)で,乳酸濃度は上昇する場合がある.CSF乳酸濃度は脳梗塞後に上昇することもある.
白質異常症. Scarpelli et al [2013], Morato et al [2014], Wu et al [2016]参照.
疫学
ミトコンドリア異常症は以前考えられていたよりも多い(表2).利用できるデータに基づくと,すべてのミトコンドリア異常症の発現頻度は推定で11.5/100,000(~1/8,500)である.Arpaらによるスペインでの推定頻度は,14歳以上で5.7/100,000である.表2. ミトコンドリア異常症の発生頻度
| 対象集団 | 病的変異もしくは疾患 | 発生頻度/100,000 (95%C.I.) 1 |
|---|---|---|
| 北イングランド2 頻度(8/97), 集団サイズ 2,122,290 |
全mtDNA欠失 | 1.333 (0.76-1.89) |
| 全mtDNA点変異 | 5.243 (4.12-6.37) |
|
| m.11778G>A & m.3460G>A (LHON) | 3.293 (2.39-4.18) |
|
| m.3243A>G | 0.953 (0.47-1.43) |
|
| m.8344A>G | 0.253 (0.01-0.5) |
|
| 全mtDNA病的変異 | 6.574 (5.30-7.83) |
|
| 北フィンランド5 成人頻度,集団サイズ 245,201 |
m.3243A>G | 5.71 (4.53-6.89) |
| 西スウェーデン6 16歳以下の子供,集団サイズ 385,616 |
小児性ミトコンドリア脳筋症 | 4.77 (2.8-7.6) |
| ヴィクトリア,オーストラリア8 出生頻度,集団サイズ 1,710,000 |
幼児性呼吸鎖疾患 | 4.79 (3.2-5.0) |
| 総括 | ミトコンドリア異常症を持つ成人と小児 | ~11.5 |
注:ミトコンドリア遺伝コードはQuick Referenceによる. 遺伝コード,遺伝構造,
- C.I.=信頼区間
- Chinnery et al [2000]
- ミトコンドリア異常症の頻度は成人発症者を基にしている(16-65歳男性,16-60歳女性)
- mtDNA変異の頻度は退職年齢(男性65歳以上,女性60歳以上)以下を対象とする.
- Majamaa et al [1998]
- Darin et al [2001]
- 1999年1月1日時点における頻度
- Skladal et al [2003]
- 1987年から1996年の出生頻度
病因
ミトコンドリア異常症は核DNAもしくはmtDNAの変異によって引き起こされる [Koopman et al 2012].ミトコンドリア異常症の分類は困難である.臨床における分類が有用とされる(表1).多くの患者は特定の疾患カテゴリーに収まらない.このようなケースは遺伝型と表現型にあまり相関が見られない複雑さにある.例として,眼筋麻痺の疾患グループは臨床上分別が付かないが,ある患者では大部分にわたるmtDNAの欠損,その他の患者にはmtDNAに点変異(例 m.3243A>G),またその他の患者では二次的なmtDNA異常を引き起こす常染色体優性核DNA変異(例 ANT1遺伝子変異)を保持していることがある.
近年におけるミトコンドリア異常症の分子生物学的研究によって,これら核遺伝子変異(表3a)とミトコンドリア遺伝子変異(表3b)による疾患の分類がより明確にされた.しかし遺伝学的分類にも次のような欠点がある.
- 現時点では大多数の患者,特に幼児期における変異を同定するのは不可能である.
- さらに,同じ変異でも大きく異なる臨床型を引き起こすことがある(例 m.3243A>G点変異は慢性進行型外眼筋麻痺(CPEO),糖尿病と難聴,または再発性脳卒中と癲癇を伴う重症の脳筋症などのいずれかを引き起こす).
- ゲノム検査法(例:エキソームシークエンス法)による新しい遺伝子発見率のため,1つのリソースで,従来知られているミトコンドリア機能を障害する全ての遺伝子リストを網羅することが難しくなっている.
表3a. ヒトミトコンドリア異常症の遺伝型分類: 核DNAの変異
| 核DNAの変異 | |
|---|---|
| ミトコンドリア呼吸鎖の核遺伝性疾患 (構造サブユニットをコードする変異遺伝子)1 |
複合体Ⅰ欠損を伴うリー症候群(NDUFS1, NDUFS2, NDUFS7, NDUFS8, NDUFV1) 複合体Ⅱ欠損を伴うリー症候群(SDHA) 複合体Ⅰ欠損を伴う白質ジストロフィー(SDHAF1) 心筋症と脳症(複合体Ⅰ欠損)(NDUFS2) 視神経萎縮と運動失調(複合体Ⅱ欠損)(SDHA) 低カリウムと乳酸アシドーシス(複合体Ⅲ欠損) (UQCRB) |
| ミトコンドリア呼吸鎖の核遺伝性疾患(集合因子をコードする変異遺伝子)1 | リー症候群(SURF1,LRPPRC) 肝炎とケトアシドーシス(SCO1) 心筋症と脳症(SCO2) 白質ジストロフィーと腎尿細管症(COX10) 過栄養心筋症(COX15) 脳症,肝不全,尿細管症(複合体Ⅲ欠損)(BCSIL) 脳症(複合体Ⅴ欠損)(ATPAF2) |
| >ミトコンドリア呼吸鎖の核遺伝性疾患(翻訳因子をコードする変異遺伝子)1 | リー症候群(GFM1) 乳酸アシドーシス,発育障害,形態異常症(MRPS16) 筋炎と鉄芽球性貧血(PUS1) 白質ジストロフィーや多小脳回症(TUFM) COX欠乏によるリー症候群と視神経萎縮(TACO1) |
| いくつかあのミトコンドリDNA欠損または欠失に関連する核遺伝性疾患 | 常染色体性進行性外眼筋麻痺(POLG, POLG2, TWNK, SLC2544) ミトコンドリア神経性胃腸管系性脳筋症(チミジンホスホリラーゼ欠損)(TYMP) Alpers-Huttenlocher syndrome (POLG) 運動失調ニューロパチー症候群2(POLG, TWNK, OPA1) 幼児(期)ミオパチー/脊髄筋萎縮症(TK2) 脳筋症と肝不全(DGUOK) 緊張低下,運動障害,そして/またはリー症候群,メチルマロン酸尿症(SUCLA2) 緊張低下,脳症,腎尿細管症,乳酸アシドーシス(RRM2B) 合成RC欠乏を伴うミトコンドリア脳症(AIF1) 可逆性肝炎(TRMU) 白内障と合成RC欠乏を伴う筋症(GFER) |
| 他の疾患 | コエンザイムQ10欠乏(COQ2,COQ9,CABC1,ETFDH) Barth症候群(メチルグルタコン酸尿症2型)(TAZ) 心筋症と乳酸アシドーシス(ミトコンドリアリン酸化キャリアー欠損)(SLC25A3) |
- Nuclear Gene-Encoded Leigh Syndrome Overview参照
- MIRAS,SCAE,SANDO,MEMSAを含む
表3b.ヒトミトコンドリア異常症の遺伝型分類: ミトコンドリアDNAの変異
| ミトコンドリアDNAの変異 | |
|---|---|
| 再構成(欠損と重複) | 慢性進行性外眼筋麻痺(CPEO) キーンズ・サイヤー症候群 糖尿病と難聴 |
| 単一遺伝子変異 | 蛋白をコードした遺伝子 レーベル(レーバー)遺伝性視神経症(LHON)(m.11778G>A, m.14484T>C, m.3460G>A) 運動失調と網膜色素変性症を伴う神経衰弱/リー症候群(m.8993T>G, m.8993T>C) |
| tRNA遺伝子1 | MELAS(m.3243A>G, m.3271T>C, m.3251A>G) MERRF(m.8344A>G, m.8356T>C) 慢性進行性外部眼筋麻痺(CPEO)(m.3243A>G, m.4274T>C) ミオパチー(m.14709T>C, m.12320A>G) 心筋症(m.3243A>G, m.12258C>A) 脳心筋症(m.1606G>A, m.10010T>C) 非症候群性感覚難聴(m.7445A>G) |
| rRNA遺伝子1 | アミノグリコシド感受性非症候群難聴(m.1555A>G) |
ついては、MITOMAP参照.変異は最近の命名ガイドラインに従ってつけられている.ヒトミトコンドリアDNAのリファレンスシークエンスはNC_012920.1(www.mitomap.org)
- ミトコンドリアの核酸位置はL鎖.
ミトコンドリア異常症の確定診断
ミトコンドリア病はどんな進行性の全身疾患でも鑑別疾患にあげるべきである.複雑な神経学的所見,もしくは単一の神経学的徴候と他の関連する病態をもつ子供においてはしばしば,ミトコンドリア疾患を十分に評価することで、疾患がわかることがある.
ミトコンドリア疾患を示す所見は,臨床的な表現型(神経学的検査を含む身体学的検査),遺伝の方式(家族歴),表現型の幅(生化学的また組織学的所見)などがある.血液サンプルから得られたDNAの分子遺伝学的検査は,確定診断に使用される.
身体所見と神経学的評価
特別な診断が疑われるときのアプローチ.
家族歴の裏付けのあるもしくは関連ある遺伝子の知られている病的変異と同じ表現型を基にした,単一遺伝子の分子遺伝子検査に合致した表現型(表1)があるならば,ミトコンドリア機能に影響する遺伝学的な疾患の診断を確立させるのは比較的単純である。
- 核DNA由来の遺伝疾患を起こす遺伝子が知られている(例えば、ミトコンドリア神経性胃腸管系脳筋症,多くの2次的欠失のある常染色体進行性外眼筋麻痺,もしくは,古典的POLG関連疾患[MIRASやAplers-Huttenlocher症候群を含む]),臨床所見は古典的で臨床家は分子遺伝学的検査を勧めるべきである.
- MELASやMERRF,レーベル遺伝性視神経症(表3b)のような古典的な母系(ミトコンドリア)遺伝ミトコンドリア病を示すとき,適切なmtDNA検査を最初に行うべきである(分子遺伝学的検査参照)
家族歴
3世代の家系図は,遺伝形式と/または診断を示唆し,また直接分子遺伝学的検査を行う手がかりとなる.
遺伝形式
ミトコンドリア遺伝病(表3b).罹患女性はすべての子供に疾患を伝え罹患男性は子供に疾患を伝えない男性と女性の罹患者の家族歴は,ミトコンドリア遺伝病を示唆する.mtDNA変異と関連した臨床徴候の幅は広く,家族歴をみると多くの多彩な症状を示す家族が含まれている(例えば,家族のうちの数人の,唯一の病状としての糖尿病もしくは軽い感覚難聴).
常染色体劣性遺伝(Table 3a).兄弟のみが罹患している家系(すなわち、家族の1世代)で,加えて/または,血族結婚である場合は常染色体劣性遺伝を示している.
常染色体優性遺伝(Table 3a).多世代で,男性も女性も罹患している家族歴は,常染色体優性遺伝を示している.はっきりした常染色体優先遺伝形式が,進行性外眼筋麻痺の患者に認められることがある.
X連鎖遺伝.罹患者が男性であり,互いに女性を経由してつながりをもつ家族歴は,X連鎖遺伝を示している.
単一の場合.もし,家族のうち一人のみ罹患がわかっているのであれば,考えられる可能性として、突然変異の場合,常染色体ミトコンドリア病と関係した病的変異の低い浸透率の場合,常染色体劣性かX連鎖疾患の家系内の一人だけの発症の場合,後天的疾患(非遺伝疾患)の場合がある.
- 進行性外眼筋麻痺かキーンズ・サイヤー症候群のほとんどの成人は単一の場合である.
- 子供時代に脳筋炎を発症した多くの患者は,両アリルの核遺伝子の病的変異かミトコンドリア遺伝子の病的変異の存在下で起きる家系内で単一の症例となる.
GeneReviewsのミトコンドリアの常染色体劣性,常染色体優性,X連鎖の遺伝形式の用語集参照.
ミトコンドリア病の疑いがあるが臨床所見が特定の疾患を示さないときのアプローチ.
病的変異が同定されないとき(特定の疾患の疑いがあるにも関わらず),もしくは臨床的な異常が複雑で,より一般的なミトコンドリア病に合致しにくいとき,非常に診断は困難である.
臨床検査はミトコンドリア病の診断を参考に行われる[Kaufman et al 2009]
表現型の確定のための他の検査
臨床所見は非特異的であるが強くミトコンドリア病を疑うとき,臨床家は血漿または脳脊髄液の乳酸濃度,ケトン体,血漿アシルカルニチン,尿中有機酸の測定を始めるべきである.
血漿/脳脊髄液の乳酸/ピルビン酸
- 血漿乳酸濃度の測定は,筋炎または中枢神経系疾患の病態を示す患者に行われる.空腹時血中乳酸濃度が3㎜/Lを超える場合は,ミトコンドリア病の診断に役立つ.
- 脳脊髄液中の乳酸濃度の測定は,中枢神経系疾患を疑う患者に行われる.空腹時脳脊髄液乳酸濃度が1.5㎜/Lを超える場合は,ミトコンドリア病の診断に役立つ
注:血漿または脳脊髄液中の乳酸濃度が基準値内でも,ミトコンドリア病の存在は否定できない.
MR分光法と運動試験 はミトコンドリア病の安静時の脳や筋肉の上昇した乳酸濃度の検出,運動後の筋肉のATPのピークの回復遅延の検出に役に立つことがある.
神経画像検査 は中枢神経系疾患を疑われる患者に行われる.CTは大脳基底核もしくは全体の萎縮を示すことがある.MRIは大脳皮質や小脳の巣状萎縮やT2強調画像で高信号の変化,特に後頭部の大脳皮質の変化を示すことがある[Scaglia et al 2005].そこには一般的な白質脳症の所見もあるかもしれない[Barragan-Campos et al 2005].小脳萎縮は小児には著明な所見である[Scaglia et al 2005].
神経生理学的検査
- 脳波は脳症またはてんかんが疑われる患者に行われる.脳症では脳波上で全体的な徐波が出現することがある.全体的または巣状の棘波や脳波の放出がてんかんの患者では認められることがある.
- 末梢神経生理学検査は四肢の衰弱,感覚症状,反射消失の患者に行われる.筋電図はしばしば正常でも筋症状を示すことがある.神経伝導速度は正常な場合でも,主に軸索の運動感覚多神経炎が認められることがある.
糖. 空腹時血糖値の上昇は糖尿病を示す場合がある.
心臓. 心電図も心エコーも両方とも,心臓病変をみつけることがある(心筋症または房室伝導欠損).
分子遺伝学的検査
分子遺伝学的診断確定はミトコンドリア病の患者のカウンセリングに重要な意味を持つ[Lieber et al 2013, Nesbitt et al 2013].例えば,乳幼児のチトクローム酸化酵素欠損は核遺伝子のSURF1またはSCOの両アリルの病的変異によって,もしくは母親由来のミトコンドリアDNAの一塩基変異(例,m.8993T>G)によって起こる.慢性進行性外眼筋麻痺は新生突然変異による欠失(例,ミトコンドリアDNAの広範囲欠失)または母系遺伝(例,ミトコンドリアDNA m.3243A>G病的変異)によって起こる.
分子遺伝学的検査と結果の解釈は複雑で,経験ある研究室,臨床専門家,遺伝カウンセラーのサポートが必要なことがある.
分子遺伝学検査は血液より採取したゲノムDNA(核DNA病的変異か複数の病的ミトコンドリアDNA変異を疑う)もしくは筋肉より採取したゲノムDNA(病的ミトコンドリアDNAを疑う)で行われる.
考えられる発端者の分子遺伝学的検査へのアプローチは単一遺伝子の一連の検査法,多遺伝子パネル検査(多遺伝子の同時検査),ゲノム検査(例:核遺伝子の病的変異を同定するためのミトコンドリアゲノムのシークエンス;エキソームシークエンスまたはゲノムシークエンス)である.
単一遺伝子または多遺伝子パネル検査
ゲノム検査に対し、一連の単一遺伝子検査と多遺伝子パネル検査は,検査するための特定の遺伝子か遺伝子セットを想定できる臨床家による.
想定は1.遺伝の形式,2.区別する臨床所見,3.ほかの鑑別できる症状 による.
病的ミトコンドリアDNA変異のための検査 は,骨格筋のDNAで通常行われる.なぜなら,病的ミトコンドリアDNA変異は血液から採取したDNAではみつからないことがあるからである.
- 広範囲PCRまたは量的PCR解析で,病的ミトコンドリアDNA再構成が認められることがある.欠失または重複の区切り点はミトコンドリアDNAシークエンスに載っていることがある.
- ミトコンドリア遺伝子のパネルを特集し病的変異を標的にした解析が行われることがある.
- 思った単一ヌクレオチドの変異が同定されなければ,全ミトコンドリアゲノムをシークエンスすることがある.
核遺伝子の病的変異の検査. 多遺伝子パネル:
- 関連した遺伝子や多遺伝子パネルに使用される方法は研究室や時間とともに違ってくる.
- 何枚かの多遺伝子パネルを使った検査法は,血液の低頻度のヘテロプラスミーのミトコンドリア変異やミトコンドリア遺伝子の欠失を検出できないことがある.それゆえ,もし病的ミトコンドリア変異による病態が疑われる場合で,複数のパネルを使った検査法は有用ではない.
ゲノム検査
もし,単一遺伝子検査(そして/もしくは多遺伝子パネルの使用)はミトコンドリア病の症状を示す患者の診断を確定できないなら,ゲノム検査を検討することもある.
注:(1)偽陰性率はゲノム領域によってさまざまである.それゆえ,ゲノム検査は標的単一遺伝子検査や多遺伝子分子遺伝学検査パネルと同じくらい正確ではないかもしれない.(2)たいていの研究室は補足の、確立された方法で陽性の結果を確認する.(3)広範囲の欠失や重複(>8-10bpの長さ),トリプレットリピート伸展,エピゲノム改変などのDNA変異によっては,ゲノム検査で検出できないかもしれない[Biesecker & Green 2014].
エキソームシークエンスは核遺伝子の変異によって起きるミトコンドリア疾患の遺伝的原理を同定するのに,非常に効果的であることがわかってきた[Taylor et al 2014].多くの呼吸鎖複合体欠損の分子学的原理を決定するために,Taylorら[2014]は次の分類に合致する53人の患者の研究を行った.
- すでに掲載された基準による呼吸鎖複合体の活性低下で確定された,臨床的に罹患した組織(骨格筋、肝臓、心臓)の組織学的そして/または生化学的なミトコンドリア疾患の診断の証拠
- 筋肉でミトコンドリアDNAのレーベルの減少が認められた患者の広範囲のミトコンドリアDNA再構成の欠失,ミトコンドリアDNAの欠失,ミトコンドリアDNAの一ヌクレオチド変異
- 先天構造異常をもつ患者の比較ゲノムハイブリダイゼーションアレイ(CGHアレイ法)による主要な各遺伝子の再構成の除外.
53人の発症者のうち,原因と推定される変異は28(53%;95%CI,39%-67%)人で同定され,疾患の可能性のある変異は4人(8%;95%CI,2%-18%)であった.全部合わせると32発症者(60%;95%CI,46-74%)になり,18の異なる遺伝子が関与していた.
他の検査(非遺伝学的検査)
多くの遺伝学的検査で診断が確定できない,または認めない患者では,疑われたミトコンドリア病の更なる検査として,呼吸鎖機能のための筋生検を含む様々な臨床検査が行われることがある.
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ミトコンドリア異常症はmtDNAもしくは核DNAの異常によって引き起こされる.
- mtDNA欠陥は母性遺伝する.
- 核遺伝子異常は常染色体劣性もしくは常染色体優性で遺伝する.
発端者の両親
- 単一mtDNA欠失
- mtDNA欠失は多くの場合新規で起こり,したがって家族内に単独で現れる.
- 単一ミトコンドリアDNA欠失の伝達は,母親からの遺伝である.
- 多発ミトコンドリア欠損を形づくる体質は,常染色体優性または常染色体劣性形式で遺伝される.
- mtDNA点変異と重複
- ミトコンドリアDNA点変異や重複は母系遺伝することがある.
- 発症者の父親はミトコンドリアDNA病的変異をもつリスクはない.
- 発端者の母親は(通常)mtDNA変異を持ち,徴候を示す場合と示さない場合がある.
発端者の同胞
- 発端者の同胞へのリスクは母親の遺伝子による. もし母親がmtDNA変異を保持しているならば,すべての同胞へ遺伝するリスクがある.
- 発端者が単一ミトコンドリア遺伝子欠損を持つとき,現在の同胞再発の推定リスクは1/24である[Chinnery et al 2004].
発端者の子
- mtDNA変異を保持する男性の子には,発病のリスクはない.
- mtDNA変異を保持するすべての女性の子は変異を遺伝するリスクがある.
- ヘテロプラスミーmtDNA点変異を持つ女性はその子孫に様々な分量のmtDNA変異を遺伝し,同じ核家族内の同胞間で大きく異なる症状を引き起こすことになる[Poulton & Turnbull 2000].
- m.8993T>G, m.8993T>C, m.3243A>G, m.8344A>G, m.11778G>AのmtDNA病的変異について,臨床的に影響する子へのリスクは母親の血液からのmtDNA変異の割合が影響することが明らかにされている[Chinnery et al 1998, White et al 1999a, Chinnery et al 2001].しかしこれらのデータは遡及的であり,遺伝カウンセリングに直接用いられるべきではない.
発端者の他の家族
その他の家族へのリスクは発端者の母親の遺伝形態による.もし母親がmtDNA変異を保持していれば,その母親の同胞とその母親もまたリスクがある.
患者家族のリスク - 常染色体劣性遺伝
発端者の両親
- 発端者の両親は絶対的にヘテロで従って対立遺伝子の一つは変異を持つ.
- ヘテロ(保因者)は無症状である.
発端者の同胞
- 概念として,発端者の同胞それぞれに発病原因となる対立遺伝子を遺伝しかつ発病する確率は25%,変異を保持する対立遺伝子を遺伝しかつ保因者となる確率は50%,正常な対立遺伝子を遺伝しかつ発病しない確率は25%である.
- リスクにある同胞が発病しないとわかった場合,その同胞が変異の保因者である可能性は2/3である.
- ヘテロ(保因者)は無症状である.
発端者の子
すべての子は絶対的にヘテロである.
患者家族のリスク - 常染色体優性遺伝
発端者の両親
- 発端者の両親は発端者と同じ病的変異を持っている場合がある.その片親は症状があることもないこともある.
- 発端者は新規の変異で罹患する場合がある.この場合,新規変異が起こる確率は不明である.
注:家族が発病しているかどうか確認できない場合,両親が発病前に死去している場合,もしくは影響する両親の発症が遅い場合など,家系の情報が有効でない場合がある.
発端者の同胞
- 同胞へのリスクは両親の遺伝形態による. 片親が病的変異を保持する場合,同胞へのリスクは50%である.
発端者の子
発端者の子それぞれに病的変異が遺伝する確率は50%である.
家族のリスク X連鎖遺伝
発端者の両親
- 罹患者の男性の父親は,疾患を持たないであろうし,病的変異の保因者でもない.
- 罹患した息子と罹患したその兄弟をもつ女性は絶対に保因者である.
発端者の兄弟姉妹.
兄弟姉妹のリスクは母親が保因者かどうかによる:
- もし,発症者の母親が病的変異を持っていれば,伝わる確率はそれぞれ50%である.病的変異を受け継いだ男性の兄弟も罹患するであろう.病的変異を受け継いだ女性の姉妹は保因者であり,通常発症しない.
- もし,発症者が単一の場合であれば(例:家族で一人の発症)であり,もし病的変異が母親の白血球DNAから発見できなかった場合,兄弟姉妹のリスクは低いが一般人口よりは高い.母親の生殖細胞系列モザイクの可能性があるからである.
発症者の子孫.
すべての罹患者の男性の娘は保因者である;彼の息子は罹患しない.
遺伝カウンセリングに関連した問題.
DNAバンキング
DNAバンクは主に白血球から調製したDNAを将来の使用のために保存しておくものである.検査法や遺伝子,変異あるいは疾患に対するわれわれの理解が進歩するかもしれないので,DNAの保存は考慮に値する.
出生前診断
ミトコンドリアDNA変異 出産前診断とその解釈はmtDNAヘテロプラスミーのため困難である.絨毛検査におけるmtDNA変異の割合は胎児組織のmtDNA変異率に反映することはなく,胎児の発育とその経過を通して変異率は変化していく[Hellebrekers et al 2012].
絨毛検査の解釈はほとんどのヘテロプラスミーmtDNA変異では困難である.
しかし,m.8993T>Gとm.8993T>Cの変異においては組織の分散とこの変異の割合は時間の経過を通してそれほど変化しない[White et al 1999b].この二つの病的変異[Harding et al 1992, White et al 1999a]に関しては,妊娠15-18週の羊水穿刺もしくは10-12週目の絨毛検査から得られる胎児のDNAによる出生前遺伝診断は有効である[Hellebrekers et al 2012].
核DNA遺伝子変異
- 分子遺伝学的検査 もし,病的変異が罹患した家族の一員で同定された場合,妊娠の増大したリスクのための出生前診断はこの遺伝子の検査かカスタム化した出生前診断を提供している臨床研究室で行われることがある.
- 生化学的遺伝学的検査 一度特定の生化学検査異常は罹患した家族のメンバーに同定されたときは,呼吸鎖複合体欠失のリスクのある妊娠のための妊娠15-18週目に通常行われる羊水穿刺から得られる羊水の培養による生化学検査によって可能である.
着床前遺伝子診断 他の家族ですでに病原変異が同定されている場合で,この方法利用できる場合がある.
関連情報
- National Library of Medicine Genetics Home Reference
- National Library of Medicine Genetics Home Reference
Mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes
- National Library of Medicine Genetics Home Reference
- United Mitochondrial Disease Foundation (UMDF)
8085 Saltsburg Road
Suite 201
Pittsburg PA 15239
Phone: 888-317-8633 (toll-free); 412-793-8077
Fax: 412-793-6477
Email: info@umdf.org
www.umdf.org
- Australian Mitochondrial Disease Foundation (AMDF)
Suite 4, Level 6, 9-13 Young Street
Sydney
Australia
Phone: 1-300-977-180
Fax: 02-9999-3474
Email: info@amdf.org.au
www.amdf.org.au
- International Foundation for Optic Nerve Disease (IFOND)
PO Box 777
Cornwall NY 12518
Phone: 845-534-7250
Fax: 845-534-7250
Email: ifond@aol.com
www.ifond.org
- Muscular Dystrophy Association - USA (MDA)
222 South Riverside Plaza
Suite 1500
Chicago IL 60606
Phone: 800-572-1717
Email: mda@mdausa.org
www.mda.org
- The Lily Foundation
31 Warren Park
Surrey CR6 9LD
United Kingdom
Phone: 07947 257247
Fax: 01883 623799
Email: liz@thelilyfoundation.org.uk
www.thelilyfoundation.org.uk
- Mitochondrial Disease Registry and Tissue Bank
Massachusetts General Hospital
185 Cambridge Street
Simches Research Building 5-238
Boston MA 02114
Phone: 617-726-5718
Fax: 617-724-9620
Email: nslate@partners.org
- RDCRN Patient Contact Registry: North American Mitochondrial Disease Consortium
臨床的マネジメント
ミトコンドリア異常症の臨床的マネジメントは主として支持的なものである[Chinnery & Turnbull 2001].ミトコンドリア異常症による不要な病苦や死を避けるため,臨床家はミトコンドリア異常に対する深い知識を持っていなければならない.
臨床的マネジメントに関わることは,初期診断,糖尿病の治療,心臓のペーシング,眼瞼下垂の矯正,白内障の眼内水晶体交換,感覚難聴おための蝸牛移植などが含まれる.
ミトコンドリア異常症の患者に様々なビタミンとコファクターが適用されている.しかし、Cochrane systematic reviewはそれらを支持するエビデンスはないことを示している[Chinnery et al 2006].
- ユビキノン(コエンザイムQ10,ユビデカレノン)としての食事サプリメントは主として許容されており,一部の患者では主観的な効果が報告されている.
- 複合体I,複合体II欠損の患者にはリボフラビンの経口投与が効果的かもしれない.
ミトコンドリア筋症における治療で,運動療法は現在評価中である[Taivassalo et al 2001, Taivassalo et al 2006, Murphy et al 2008].
コエンザイムQ10は特に,コエンザイムQ10 生合成の欠損の患者に行われている.
イデベノンはレーベル(レーバー)遺伝性視神経萎縮症 (LHON)の治療法として有用かもしれない.
ミトコンドリア機能低下の2次的原因の原因,エチルマロン酸尿は特別な治療法をすることがある[Tiranti et al 2009].
ミトコンドリア神経性胃腸管系脳筋症(MNGIE)の患者は造血幹細胞移植が効果があることがある.
検査的予防戦略
病的ミトコンドリアDNA変異の予防治療としての核移植の可能性は現在研究中である[Craven et al 2010].
最近のレビューによると,それらの使用を支持する根拠の欠落が指摘されている.食事サプリメント(例としてコエンザイムQ10やユビカドレノン)は主として許容されており,一部の患者では主観的な効果が報告されている.複合体I,複合体II欠損の患者にはリボフラビンの経口投与が効果的かもしれない.
ミトコンドリア筋症における治療で,運動療法は現在評価中である.
更新履歴
- Gene Review著者: Patrick F Chinnery, MBBS, PhD, MRCP
日本語訳者: 牧野聖子(Massachusetts General Hospital, Cardiovascular Research Center)
GeneReview 最終更新日: 2006.2.21. 日本語訳最終更新日: 2007.3.29 - Gene Reviews著者: Patrick F Chinnery, BMedSci, MBBS, PhD, FRCPath, FRCP, FMedSci
日本語訳者: 和泉 賢一 (札幌医科大学医学部遺伝医学,NGSDプロジェクト)
Gene Reviews 最終更新日: 2014.8.14 日本語訳最終更新日: 2017.12.29(in present)