フォンヴィレブランド病
(von Willebrand Disease)
[Synonyms: von Willbrand Factor Deficiency]
Gene Review著者: Anne C Goodeve, PhD
日本語訳者:江田肖(瀬戸病院遺伝診療科),櫻井晃洋(札幌医科大学附属病院遺伝子診療室)
Gene Review 最終更新日: 2014.7.24. 日本語訳最終更新日: 2016.11.16.
要約
疾患の特徴フォンウィルブランド病(VWD)は血漿フォンウィルブランド因子(VWF)の異常や欠損が原因とする先天性出血障害である。明らかな症状は止血困難だけであるため、年齢が長ずるにつれて出血歴がより明らかとなる。
1型VWD(全体の約70%)は、一般的に軽度の皮膚粘膜出血症状を示す。
2型VWD(全体の約25%)は下記のサブタイプがある。
- 2A型は、軽度から中度の皮膚粘膜出血症状を示す。
- 2B型は一般的に軽度から中度の皮膚粘膜出血症状を示すが、ある一定の状況において、血小板減少症の増悪を伴う。
- 2M型は一般的に軽度から中度の皮膚粘膜出血症状をしめす。
- 2N型は手術時の出血多量を起こし、症状は血友病Aに類似する。
3型VWD(全体の5%以下)は重度の皮膚粘膜出血及び筋骨格出血の症状を示す。
診断・検査VWDの診断は、一般的にVWDに特異的な止血因子検査及び/または唯一の原因遺伝子であるフォンウィルブランド因子(VWF)の分子遺伝学的検査が必要となる。ほとんどの場合、診断には家族歴の存在が必要である。
臨床的マネジメント
症状に対する治療:
出血性疾患に対する包括的治療はVWD患者に有効である。重度な出血症状に対しては、VWF及び第Ⅷ因子を含む血漿由来の不活性凝固因子濃縮物の静脈内注入によって予防やコントロールができる。VWDのタイプによるが、軽度な出血症状には、通常バソプレッシンアナログであるデスモプレッシンの静脈内注射もしくは皮下注射が有効である。フィブリン溶解阻害剤や月経過多に対するホルモン剤などの方法も症状緩和に有用である。VWD妊婦は、出産時もしくは産後に出血性合併症を起こすリスクが高い。
一次症状の予防:
3型VWD患者には濃縮VWF/第Ⅷ因子製剤を予防的に投与する。
二次合併症の予防:デスモプレッシンを慎重に投与する。幼児は水分摂取を制限することが困難なため、特に2歳未満の幼児において注意が必要である(水中毒を惹起しやすい-訳者注)。A型及びB型肝炎のワクチンを接種する。
サーベイランス:
出血性疾患のマネジメントに経験のある医療機関で定期検診を受ける。3型VWD患者に対しては、理学療法士による関節可動性の評価を定期的に行う。
回避すべき薬剤や環境:外傷のリスク、特に頭部外傷のリスクを伴う活動は避ける。血小板機能に影響を及ぼす薬剤(アセチルサリチル酸、クロピドグレル、非ステロイド系抗炎症薬)の使用を避ける。男児の割礼は血液専門医との相談の後に行われるべきである。
リスクのある血縁者における評価:
家系内における病原性変異が同定されている場合、リスクのある血縁者に対する分子遺伝学的検査によって、早期診断及び早期治療(必要であれば)が可能となる。
妊娠中の管理:
VWF値は妊娠中に増加するため、ベースラインVWFと第Ⅷ因子値が30IU/dL以上の女性患者は分娩まで正常値になることが多い。しかし、基礎値が20IU/dL以下もしくは、ベースラインVWF:RCo/VWF:Ag比が0.6以下の患者には代替療法が必要となる。デスモプレッシンは1型VWD女性患者の分娩時出血のコントロールに有効であるが、遅れてくる産後の二次出血が問題となる。
研究中の治療:
遺伝子組み換えVWFは、現在臨床試験中であり、血漿由来VWFに代わって使用されると見込まれている。
遺伝カウンセリング
ほとんどの1型、ほとんどの2A型、全ての2B型及び全ての2M型は、常染色体優性遺伝の形式をとる。2N型、3型及びいくつかの1型と2型は、常染色体劣性遺伝の形式をとる。
- 常染色体優性遺伝:ほとんどの患者は、罹患している親を持つ。突然変異の割合は分かっていない。常染色体優性VWD患者の子は50%の確率で病原性変異を受け継ぐ。
- 常染色体劣性遺伝:患者の同胞は25%の確率で患者となり、50%の確率で無症候性保因者、残り25%は罹患者でも保因者でもない。家系内における病原性変異が同定されている場合、リスクのある血縁者の保因者検査は可能である。
家系内における病原性変異が同定されている場合、もしくは遺伝子マーカーを用いた連鎖解析が有用な場合、出生前診断(ほとんどが3型VWDに対し)は可能である。
GeneReviewの範囲
フォンウィルブランド病は下記の疾患を含む |
|---|
|
診断
臨床診断
フォンウィルブランド病(VWD)は、血漿中フォンウィルブランド因子(VWF)の欠失もしくは機能障害を原因とする。VWFは、血小板による止血機能を仲介し、血液凝固第Ⅷ因子を安定化させることによって一次止血における中心的な役割を果たす巨大な多量体糖タンパクである。
VWDの3つの型は[Sadler et al 2006]、
- 1型、正常なVWFの部分的量的欠乏。
- 2型、機能障害のあるVWFの質的欠失。2型はVWFの機能障害によって、2A、2B、2Mと2Nとの4つのサブタイプに分けられる。
- 3型、VWFの完全欠失(VWFは実質的に存在しない)。
下記を含む過度の皮膚粘膜出血が見られる場合、VWDが疑われる。
- 明らかな外傷を伴わないあざ
- 長引くもしくは繰り返す鼻血
- 歯磨きもしくはデンタルフロス後におこる歯肉からの出血、歯磨きもしくは抜歯後の長引く出血
- 月経過多、特に初潮以降つづく月経過多
- 外科手術、外傷、分娩後の長引く出血
VWD診断の一部として症状の発生及びその重症度を点数化する標準臨床評価ツール[Tosetto et al 2006, Bowman et al 2008, Bowman et al 2009, Rodeghiero et al 2010]の有用性が認められている。これらのツールは、出血が一般集団に比べて多いかどうか、診断の正確さ、重症度の定量化および臨床的介入の必要性を決定することができる。また、出血性疾患の疑いを否定するにもこれらのツールが用いられている[Tosetto et al 2011]。
診断にはVWDに特異的な止血因子検査(検査の項を参照))及び/またはVWF遺伝子の分子遺伝学的検査が必要である。
診断にはさらに(ほとんどの場合)家族歴の存在が必要である。注意:軽度な1型VWDでは、浸透率が不完全であることや表現型の違いによって、家族歴が認められない場合もある。
検査
スクリーニング検査
- 全血球計算(CBC)は正常であっても、特に2B型VWDにおいて、小球性貧血(患者が鉄欠乏性貧血である場合)や血小板数の低下(血小板減少症)を示すことがある。
- 活性化部分トロンボプラスチン時間(aPTT)は通常正常である。ただし、重症の1型VWD、2N型VWD、3型VWDで見られるように、第Ⅷ因子が30~40IU/dLを下回った場合、aPTTは延長することがある。第Ⅷ因子凝固活性の正常範囲は約50~150IU/dLである。
- プロトロンビン時間(PT)は正常である。
- その他 VWD疑いの患者に対し、皮膚出血時間や血小板機能分析(PFA閉止時間)を行う検査機関もあるが、出血異常が軽度な患者では、これらの検査は感度は低い。
止血因子検査 スクリーニング検査では正常であっても、下記の止血因子検査(Table 1)は必ず実施する[Budde et al 2006]。注意:検査値の正常範囲は検査機関ごとに決められているため、検査結果は1つの指標にすぎない。
- VWF:Rco 機能的VWF検査(リストセチン補因子活性検査とも呼ばれる)は抗生物質リストセチンによって誘発されるVWFの血小板凝集能を調べる検査である(正常範囲は約50~200IU/dL)。いくつかの新しいVWF活性検査は利用可能になったが、VWF:RCoとの比較では、これらの使用を推奨するにはまだ十分ではない。
- VWF:Ag 酵素結合免疫吸着検査(ELISA)もしくはラテックス免疫検査(LIA)を用いて、血漿中VWFタンパク(抗原)量(正常範囲は約50~200IU/dL)を測定する[Castaman et al 2010a]。VWF:RCo/VWF:Ag比の低下(<0.6)は、高分子量(HMW)多量体の欠損を示唆する。
- 第Ⅷ因子活性レベル 機能的第Ⅷ因子検査(例:凝固カスケードにおける第Ⅷ因子の活性)(正常範囲は約50~150IU/dL)。
上記3つの検査で異常が認められた場合、VWDのサブタイプを特定するために、血液凝固の専門施設は次の検査を行う。
- VWF多量体分析ではSDSアガロースゲル電気泳動を用いて、血漿中のVWFオリゴマーの補因子を決定する。正常な血漿中には、二量体から40を超える多量体VWFが含まれている。これらの多量体は低分子量(1~5量体)、中分子量(6~10量体)と高分子量(10量体以上)のものに分類される。2A型及び2B型VWDでは、高分子量(HMW)多量体が減少かもしくは欠損する。2A型VWDでは、さらに中分子量多量体が欠損する場合もある。サテライト("トリプレット")バンドパターンの異常は、2型VWDの発症病理や分類に有用である[Budde et al 2008]。
- リストセチン誘導血小板凝集(RIPA)検査 では2~3倍濃縮リストセチンを用いて、VWFの血小板凝集能を調べる。低濃度(~0.5-0.7mg/mL)のリストセチンでも凝集すれば、VWFと血小板の結合が増強する、2B型VWDもしくはGPIBA遺伝子病原性変異が原因の血小板型VWD(PT-VWD)(鑑別診断の項を参照)となる。
- VWFによる第Ⅷ因子結合検査(VWF:FⅧB)はVWFの第VIII凝固因子への結合能検査である。2N型VWDを同定するために必須である。
- コラーゲン結合検査(VWF:CB)はVWFのコラーゲン(血管内皮下マトリックスの成分)への結合能検査である。機能障害のあるVWF(例えば、1型VWDと2型VWDの鑑別)を明確にするために行われている[Flood et al 2013]。コラーゲンI/III混合物はよく使われるが、最近コラーゲンVIへの単独結合欠損が確認された[Flood et al 2012]。検査値の正常範囲は約50~200IU/dLである。VWF:CB/VWF:Ag比の低下は高分子量(HMW)多量体の欠損を示唆する。
Table1
特異的VWF検査に基づくVWDの分類
VWDの型 |
VWF:Rco1 | VWF:Ag1 | Rco/Ag | 第Ⅷ因子:C IU/dL1 |
多量体 パターン2 |
その他 |
|---|---|---|---|---|---|---|
| 1 | 低値 | 低値 | 等値 | ~1.5×VWF:Ag | 基本的に正常 | |
| 2A | 低値 | 低値 | VWF:RCo<VWF:Ag | 低下もしくは正常 | 異常 ↓高分子量 |
|
| 2B | 低値 | 低値 | VWF:RCo<VWF:Ag | 低下もしくは正常 | 通常は異常 ↓高分子量 |
↑RIPA3 (↓血小板数) |
| 2M | 低値 | 低値 | VWF:RCo<<VWF:Ag | 低下もしくは正常 | 高分子量の欠損 を伴わない |
|
| 2N | 正常/低値 | 正常/低値 | 等量 | <40 | ほとんどは正常 | ↓VWF:第Ⅷ因子4 |
| 3 | 欠損 | 欠損 | NA | <10 | 欠損 |
- 参考値(概算値)との比較 VWF:RCo(50~200IU/dL)、VWF:Ag(50~200IU/dL)、第Ⅷ因子活性レベル(50~150IU/dL)
- 高分子量(HMW)多量体
- 凝集の増加は低濃度のリストセチンでも見られる
- VWFの第Ⅷ因子への結合能および保護能が低下する。VWFと第Ⅷ因子の値は、中度の血友病Aの男性患者や症候性血友病A保因者女性の値と非常に似ている。
分子遺伝学的検査
遺伝子
VWF遺伝子は病原性変異がVWDを引き起こすことが知られている唯一の遺伝子である。
注:
- 現行の分類法では、VWDがVWF遺伝子における病原性変異が同定できるものとは限らない。いくつかの「明らかな」VWD症例において、VWFコード領域の病原性変異が同定されていない。サンガー配列解析に加えて量的解析を用いた最近の研究では、3型VWD(n=18)の94%と2型VWD(n=32)の94%において、病原性変異が同定された。しかし、1型VWD(n=28)と診断された患者において、病原性変異が同定されたのは68%しかなかった[Yadegari et al 2012]。この研究における病原性変異の検出率はほかのいくつかの研究に近い。VWFに病原性変異が同定されなくても、VWDから除外することができない。
- 血小板型偽性VWD(PT-VWD)はGPIBAにおける病原性変異が原因であるが、2B型VWDのような表現型を示す(鑑別診断の項を参照)。
臨床的検査 VWFドメイン構造及び各ドメインをコードするエクソンをFigure 1に示す。
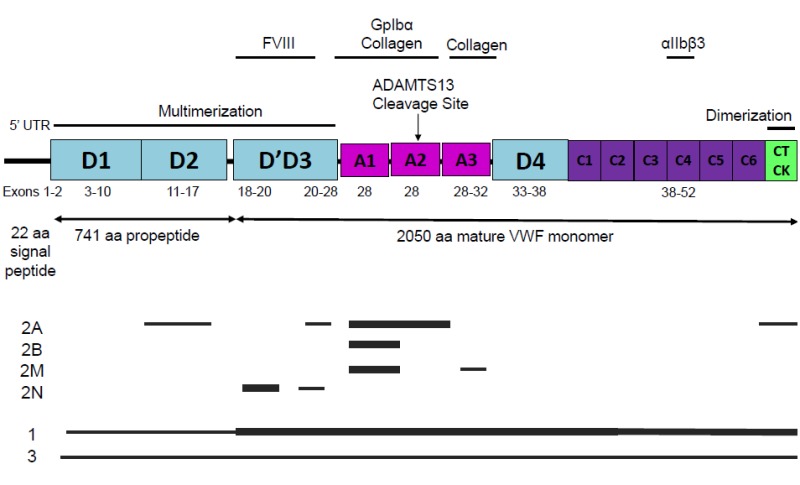
Figure 1
VFWタンパク構造[Zhou et al 2012より引用]および各型のVWDによるVWF病原性変異の分布。太線は病原性変異の頻度が最も高いエクソン、細線は比較的に頻度が低いエクソンの場所を示している。2型VWDにおいて、VWFの機能およびドメイン領域に影響を与える病原性変異は主にミスセンス変異である。
- 遺伝子配列分析 VWF遺伝子全コード領域およびエクソン/イントロン隣接領域の配列分析は可能である。ただし、遺伝子のサイズ、構造および一部の偽遺伝子によって、解析は複雑になる(分子遺伝学の項を参照)。
- 連鎖解析 3型VWDの患者において、2つの変異アレルを同定できない場合、連鎖解析は出生前診断に有用である。VWFにおける正常なアレル変化は連鎖解析に有用であり、Table 4を参照する。
- 欠失/重複解析 コピー数の変化を検出するすべての検査法によるVWFの欠失/重複解析は可能である[Yadegari et al 2011]。しかし、VWD患者における大規模な欠失や重複の割合はまだ確立されていない。また、以前には大規模なヘテロ接合欠失変異は見逃されていた可能性がある。最近、ISTH-SSC VWF Databaseに登録されている3型VWD患者の約7%は大規模な欠失によるものと報告されている。大規模なin-frame欠失は1型と2型VWDにも報告されている[Sutherland et al 2009, Casari et al 2010]。また、1つもしくは2つのエクソンにおける二か所の重複は学会抄録に報告されている[Schneppenheim et al 2011, Boisseau et al 2013]。
- 遺伝子転換 はイントロン27の3'末端からエクソン28の5'末端までの配列に影響を与える。病原性変異は、2箇所もしくはそれ以上の偽遺伝子配列(VWFP)由来の連続した配列変異をコード遺伝子に置き換えることで確認できる。6-335bpの変換が最も多く見られ、1型、2B型および3型VWDに報告されている。
1型VWD 1型VWD患者の60~65%は病原性変異が同定されている[Cumming et al 2006, Goodeve et al 2007, James et al 2007a, Yadegari et al 2012]。
- VWF:AgとVWF:RCoレベルが25IU/dL未満の時、通常完全浸透で優性遺伝形式のミスセンス変異が確認できる。
- VWF:AgとVWF:Rcoレベルが25~50IU/dLの患者の約50%で、p.Tyr1584Cysのような不完全浸透で優性遺伝形式のミスセンス変異が確認されている。
- VWF値が35IU/dL以上の患者において、不完全浸透なVWF病原性変異がどの程度表現型(出血)に関与しているかは明らかではないため、遺伝子検査結果の解釈は容易でない[Keeney et al 2008]。
1型VWDにおける病原性変異の約50%がエクソン18から28の間に存在するので、これらのエクソンを先に解析する。しかしながら、全ての変異を確認するためには、全遺伝子配列を解析する必要がある。
2型VWD 2A型や2M型のほとんどの病原性変異および2B型のすべての病的ミスセンス変異はエクソン28に存在する。従って、この3つのサブタイプを疑う場合は、エクソン28を最初に調べるべきである。
- 常染色体優性2A型VWD ほとんどの病原性変異はエクソン28に存在し、A2およびA1ドメイン(A2より少ない)に影響を与える。D3集合体(エクソン22およびエクソン25-27)における病原性変異(常染色体優性)もよく見られる[Schneppenheim et al 2010]。
- 2A型VWD エクソン11-16(常染色体劣性)とエクソン52(常染色体優性と常染色体劣性)におけるミスセンス変異も報告されているため、これらのエクソンは次に調べるべきである。さらに、ほかのいくつかのエクソンに存在する病原性変異に関する症例報告がある。
- 常染色体劣性2A型VWD 罹患者は同じミスセンス変異のホモ接合型(近親婚の家系で良く見られる)か、もしくはミスセンス変異とnull-alleleとの複合接合型かのどちらかである。
- 常染色体優性2B型VWD ミスセンス変異はエクソン28もしくはA1ドメインの近接領域に存在する[Federici et al 2009]。
- 常染色体優性2M型VWD ほとんどの病原性変異はエクソン28に存在する。一部のコラーゲン結合異常を引き起こす変異は、エクソン29-32がコードするA3ドメインに存在している[Keeling et al 2012]。
- 常染色体劣性2N型VWD ほとんどのミスセンス変異はエクソン18-20に存在する。またかなり低い割合ではあるが、エクソン17と24-25に存在するという報告もある[Mazurier & Hilbert 2005]。
- ミスセンス変異のホモ接合は一定の割合で見られる。特にp.Arg584Glnのヘテロ接合は北欧集団の1%を占める。
- 患者の多くはミスセンス変異とnull-alleleを生じる病原性変異の複合ヘテロ接合型である。また、頻度は多くないが、2つのミスセンス変異をもつ複合ヘテロ接合型の患者もいる。
3型VWD 3型VWDに関与する病原性変異は、VWFコード領域全体のいたるところ(例えば、エクソン2-52)に見られる。コード領域全体の遺伝子配列分析に加えた欠失/重複解析は約90%の3型VWDにおける病原性変異を同定できる。
- 20%はミスセンス変異である。変異はD1(エクソン3~10)及びA1-A2(エクソン28)にも存在する(Figure 1)。
- 80%はnull-alleleであり、VWF遺伝子の全領域に見られる(Table A)。null-alleleは様々な異なるタイプの変異によって生じる。mRNA、タンパクの完全欠損/不安定性、もしくは非機能的遺伝子産物の発現によって、null-alleleは機能的なタンパク産物を生成できない(例:分泌異常のタンパク)。
- 欠失/重複(遺伝子量)解析の利用は可能であるが、VWDにおける大規模な欠失/重複の有効性はまだ確立されていない。ISTH-SSC VWF Databaseに登録されている3型VWD患者において、大規模な欠失は約7%を占めている。
- 出生前診断を希望する3型VWDの家系において、2つのアレルにおける病原性変異が同定されていなければ、連鎖解析は有用である。
Table 2
フォンウィルブランド病(VWD)における分子遺伝学的検査の概要
| 遺伝子1 | 型 | VWDに占める割合 | 検査方法 | 検出される変異 | 発端者における病原性変異の検出率 |
|---|---|---|---|---|---|
| VWF | 1型 | ~70% | 全コード領域および隣接するイントロンとの境界領域の遺伝子配列解析 | 配列変異3 | 60~65% |
| 欠失/重複解析 | 部分および全遺伝子欠失/重複 | <5% | |||
| 選択的エクソン配列解析 | エクソン18-28の遺伝子配列解析 3 | ~50% | |||
| 2型すべて | ~25%5 | 選択的エクソン配列解析 | 配列変異 3 | ~90%6 | |
| 2A型(AD)、2B型、2M型 | 脚注2を参照 | 選択的エクソン配列解析 | エクソン28の配列変異 3 | ~70% | |
| 2A型(AR) | 脚注2を参照 | エクソン11-16、22、25-27及び52の配列変異 3 | 2A型患者において、 ~30% |
||
| 2N型 | 脚注2を参照 | エクソン18-20の配列変異3 | ~80% | ||
| 3型 | <5%7 | 全コード領域およびイントロンとの隣接領域の遺伝子配列解析 | 配列解析 3 | ~90% | |
| 欠失/重複解析4 | 部分および全遺伝子欠失/重複 | <10% | |||
| すべての型 | NA | 連鎖解析 | NA | NA |
AD=常染色体優性遺伝形式
AR=常染色体劣性遺伝形式
NA=適用なし
- 染色体座位はTable A.遺伝子とデータベースの項を参照する。アレル変異に関する情報は分子遺伝学の項を参照する。
- 検査法の特定の遺伝子における変異を検出する能力。
- 遺伝子配列分析によって検出される病原性変異は、小規模な遺伝子内欠失/挿入、およびミスセンス、ナンセンス、スプライシングサイト変異を含む。一般的に、エクソンもしくは全遺伝子欠失/重複は検出されない。遺伝子配列分析の結果に関する解釈は、ここを参照とのこと。
- 遺伝子配列分析では検出できない、ゲノムDNAのコード領域および隣接するイントロンの欠失/重複を同定する検査法は、この遺伝子/染色体領域を含む定量的PCR、long-range PCR、MLPAまたは染色体マイクロアレイ(CMA)がある。
- 2型VWDはVWD全体の約25%を占める。ヨーロッパ起源の集団において、各サブタイプの割合は2A>2M>2N>2Bである。
- Yadegari et al [2012]
- 近親婚の多い集団において、劣性遺伝形式のVWDの割合は上昇し、3型VWDは罹患者の大部分を占める。
検査の特徴 検査感度および特異度を含めた検査の特徴に関する情報はClinical Utility Gene Card [Cumming et al 2011]を参照する。
検査手順
発端者における診断の確定/確立.2型VWDの患者において、分子遺伝学的検査よりも、特異的VWD止血因子検査から明確な診断が得られる。
VWDに対する分子遺伝学的検査は通常、下記の場合において実施される。
- VWDのサブタイプの確立。特異的VWD止血因子検査からVWDを示唆された患者において、遺伝学的検査からはより確定的診断を得られる。
- 表現型の検査だけでは判断できない2N型VWD、軽症の血友病A(男性)もしくは症候性血友病Aの保因者(女性)の鑑別(例えば、VWF:FⅧB因子検査は利用できないもしくは判断できない)。
- 治療法の異なる2B型VWDとPT-VWDの鑑別。
- 3型VWDの家族における出生前診断。
- 家族歴から遺伝形式が明確ではない家系において、遺伝形式の決定および血縁者のリスク評価のため、分子遺伝学的検査は実施される。
VWD遺伝学的検査のガイドラインは、英国血友病センター医師団体(UK haemophilia centre doctors organization)より公開されている[Keeney et al 2008]。
臨床的特徴
臨床像
VWDは出血傾向を特徴とする先天性疾患である。症状は出血が止まらないことで気付かれ、出血歴は年齢とともに明白となる。したがって出血傾向が明らかになるまで多少時間がかかることがある。
出血歴は疾患の重症度に依存する。3型VWDは早い時期に明らかになるが、一方で1型VWDは、出血の既往があったとしても、中年期まで診断されないことがある。
VWDの患者は過度の皮膚粘膜出血(あざ、鼻出血、月経過多など)を起こす。しかし、2N型や3N型において、第Ⅷ因子活性が10IU/dLよりも低い場合は、筋骨格出血が生じうる。
1型VWD は一般集団(非近親婚集団)におけるVWDの約70%を占める。1型VWDは一般的に軽度の皮膚粘膜出血症状をしめす。しかしVWF値が15IU/dLを下回ったときには重症化する。鼻血やあざは小児に共通の症状であり、月経過多は生殖年齢の女性に最もよく見られる[James & Lillicrap 2006, Kadir & Chi 2006, Tosetto et al 2006]。
2型VWDはすべてのVWDの約25%を占める。ヨーロッパ集団におけるサブタイプの相対頻度は2A>2M>2N>2Bの順である。
- 2A型VWD 通常、軽度から中度の皮膚粘膜出血症状をしめす。
- 2B型VWD 通常、軽度から中度の皮膚粘膜出血症状をしめす。血小板減少症をしめすこともある。2B型VWDに顕著な特徴は、重症感染症や外科手術、妊娠などストレスの多い状況もしくはデスモプレッシンを投与したときに血小板減少症が悪化することである。
- 2M型VWD 通常、軽度から中度の皮膚粘膜出血症状をしめす。しかしVWF:RCo値が極端に低いもしくは0の場合、出血は重症化する[Castaman et al 2012]。
- 2N型VWD 基本的に中度の血友病Aと同じ症状を示す。この2つの疾患はともに第Ⅷ因子活性の低下が原因であり、手術や外科的処置の時に出血過多が起こる。
3型VWD は近親婚の頻度の高い地域を除き、全VWDの5%未満である。過度の皮膚粘膜出血や筋骨格出血の両方を含む重度な出血症状を示す[Metjian et al 2009]。
遺伝子型と臨床型の関連
一般的に、VWF値と出血の重症度との間には反比例の関係がある[Tosetto et al 2006]。出血スコアはいくつかのコホート研究に記録され、異なるVWDおよび特定な病原性変異に関連した出血の範囲を示す指標となる。
Table 3 VWFのタイプによる出血スコア
| 患者群 | 研究 | 患者数 | 出血スコア中間値 | 出血スコア範囲 |
|---|---|---|---|---|
| 1型 | Goodeve et al [2007] | 150 | 9 | -1-24 |
| 2A型 | Castaman et al [2012] | 46 | 11 | 6-16 |
| 2B型 | Federici et al [2009] | 40 | 5 | 4-24 |
| 2M型 | Castaman et al [2012] | 61 | 7 | 4-28 |
| 3型 | Solimando et al [2012] | 9 | 15 | 6-26 |
| 3型 | Bowman et al [2013] | 42 | 13 | 3-30 |
出血スコアが高ければ高いほど、出血が重症である。
注:上記の研究では同様な出血評価ツールを使用されていたが、各ツールおよびそれらの使用方法におけるわずかな違いはそれぞれの出血スコアに影響を与える可能性がある。
2N型VWD ミスセンス変異によって、VWFの持つ第Ⅷ因子への結合能及び保護能が低下する。VWFと第Ⅷ因子の値は、軽症の血友病A男性患者と症候性の血友病A女性保因者と全く同じに見える。
ABO血液型 血液型によって血漿中VWF値において約25%の相違が見られる。VWFのABOグリコシル化は、クリアランスの割合に影響を与える[Jenkins & O'Donnell 2006]。非O型患者はO型患者よりVWF値が高い。AB型患者のVWF値は最も高い。共通の病原性変異p.Tyr1584Cysを持つ1型VWDにおいて[O'Brien et al 2003, Davies et al 2007]、ABO血液型は浸透率及びVWF低値に重要な因子である[Goodeve et al 2007, James et al 2007a]。
浸透率
常染色体優性1型VWD 血漿VWFが25IU/dL以下になるような病原性変異の浸透率はほぼ完全である。血漿VWFが25IU/dL以上の場合、通常浸透率は不完全である。
他の常染色体優性遺伝形式の2A型、2B型と2M型の浸透率は通常完全である。
命名
命名の変更は下記となる
- von Willebrand's diseaseは、von Willebrand diseaseに変更
- vWFはVWFに変更。
- vWDはVWDに変更
- RiCof(リストセチン補因子活性)は、VWF:RCoに変更[Mazurier & Rodeghiero 2001]。命名はISTH VWF Web siteを参照ください。
- FⅧ RAg(第Ⅷ因子関連抗原)はVWF:Agに変更。
- 血小板型偽性VWD(Platelet-type pseudo von Willebrand disease, PT-VWD)は、偽性VWDとも呼ばれ、GP1BA遺伝子の病原性変異が原因である。このためVWDに属さない(鑑別診断の項を参照)。
- 後天性フォンウィルブランド症候群(Acquired von Willebrand syndrome:AVWS)は後天性VWDに代わって、専門用語として推奨されている。AVWSで見られるVWFの凝集、構造および機能の欠陥は、ほかの医学的状態の結果として生じ、遺伝性のものでもなく、VWF遺伝子の変異によるものでもない(詳細は鑑別診断のAVWSを参照)。
頻度
VWD患者は人口の0.1%から1%を占める。1万人に1人は専門機関への紹介が必要である。
3型VWDは100万人に0.5~6人が罹患するが、近親婚の割合とともに上昇する。
遺伝学的関連(アレル)疾患
本章に記述された疾患のほかに、VWF病原性変異による表現型は知られていない。
鑑別診断
以下の2つの疾患は、表現型からVWDとの鑑別が難しい。
- 軽症型血友病AはF8病原性変異が原因で、第Ⅷ因子活性の低下(~5-40IU/dL)および正常から境界型のVWF低値が見られるため、2N型VWDと類似する。VWFの第Ⅷ因子への結合能を決定するVWF:第Ⅷ因子検査はこの両疾患の鑑別に用いられる[Casonato et al 2007]、市販の検査も利用できるようになった[Veyradier et al 2011]。しかし、この検査の有効性は限られている場合がある。代わりに、分子遺伝学的検査はこの両疾患の鑑別に利用できる。
第Ⅷ因子活性の低下が見られる家系においては、X連鎖の遺伝形式が軽症の血友病Aとの鑑別に有用である。F8病原性変異に加え、偏ったX染色体不活化が原因であることが多いため、家族歴からの情報が不十分である場合は、たとえ孤発の症候性の女性(家族内で単一の発症)であっても、F8の遺伝子配列分析を最初に行うべきである。これらの症例ではF8染色体内逆位が考えられ、DNA配列解析もしくはF8遺伝子のエクソン1-26解析を行うべきである。女性ではヘテロ接合の部分的もしくは完全な遺伝子欠損/重複を同定するために、F8遺伝子の量的解析を用いる。2N型VWDもしくは血友病Aの疑いで検査した患者のうち50%以上で病原性変異が見つかっている。F8遺伝子に病原性変異がなかった場合、VWF遺伝子エクソンのスクリーニングを行う。
- PT-VWD(偽性VWDとも呼ばれる)(OMIM 177820)はGP1BA遺伝子における病原性変異が原因で発症し、2B型VWDとの鑑別が難しい。患者/コントロールの血漿と血小板を混和し、どの血液内成分に異常があるかを決定することで、この2つの疾患の鑑別が可能である[Favaloro et al 2007, Favaloro 2008, Franchini et al 2008]。VWFエクソン28に病原性変異がない場合は、GP1BA遺伝子のエクソン2における病原性変異の有無を確認する。今まで、GpIbαアミノ酸Trp256とMet255の間にあるミスセンスを加え、Pro449からSer457までの27bpのin-frame欠失(1345_1371del27)[Othman et al 2005, Hamilton et al 2011, Woods et al 2014, PT-VWD Registry]が報告されている(すべての標準命名法はHuman Genome Variation Societyに従う。参考配列はNP_000164.4とNM_000173.4である)。
PT-VWDは過小診断される可能性が高い。また、誤診は効果のない治療につながる。VWF濃縮物は低下したVWFを補正するのに必要である。血小板減少が著しい場合は、血小板輸血が必要とある。異常なGpIbαへの結合が原因で、置換したVWFの半減期は低下する。したがってVWF濃縮物はVWDのときよりも頻回に投与する必要がある。2B型VWDと診断された患者の約15%は、分子遺伝学的検査によって、GP1BA遺伝子におけるミスセンス変異もしくはin-frame変異を同定できる[Hamilton et al 2011]。
後天性フォンウィルブランド症候群(AVWS)は軽度から中度の出血傾向を示す疾患で、様々な状況で起こるが[Federici 2006, Nichols et al 2008, Sucker et al 2009, Federici et al 2013]、VWF遺伝子変異がその原因ではない。AVWSは40歳以上で出血歴がない人によく見られる。AVWSの発症原因は様々であり、以下のようなものがある。
- リンパ増殖性疾患や形質細胞疾患、異常タンパク症(意義不明の単クローン性高γグロブリン血症[MGUS])、多発性骨髄腫、Waldenstromマクログロブリン血症。これらの疾患のいくつかでは、VWFに対する自己抗体が見られる。
- 全身性エリテマトーデス(SLE)や強皮症、抗リン脂質抗体症候群を含む自己免疫疾患。
- VWFタンパク分解の上昇を来たすようなせん断誘起VWF構造変化(例えば大動脈弁狭窄、心室中隔欠損など)。
- 血小板数の著増(例えば本態性血小板血症やその他の骨髄増殖性疾患)
- 腫瘍細胞への異常な結合によって、VWFは血液循環中から除去される(例えばウィルムス腫瘍やいくつかのリンパ増殖性疾患)
- VWF合成の低下(例えば甲状腺機能低下症)
- 特定の薬剤(例えばバルプロ酸、シプロフロキサシン、グリセオフルビン、ヒドキシエチルスターチ)
臨床的マネジメント
最初の診断確定後の評価
VWDと診断された患者の症状の程度および治療の必要性を確認するために以下の評価方法が推奨される。
- 本人の既往および家族歴は重症度予測及び治療選択に有用である。出血評価ツールを使用することで、標準的評価の確立は容易である[International Society on Thrombosis and Haemostasis 2011, Tosetto et al 2011]。
- 3型VWD患者に対する関節及び筋肉の評価(1型及び2型VWDでは筋骨格出血は稀)。
- 3型VWDと診断された場合や1985年以前に血液製剤もしくは血漿由来凝固因子治療を受けた場合は、B型肝炎、C型肝炎及びHIVのスクリーニングを行う。
- VWD患者の多く、特に月経過多の女性は鉄が欠乏しているので、血清鉄濃度とフェリチン濃度(鉄貯蔵量を調べるため)の基準値を調べる。
- 月経過多の女性に対する婦人科的評価[Demers et al 2005]
- 臨床遺伝相談
症状の治療
治療ガイドラインはNichols et al [2008] (full text)とCastaman et al [2013] (full text)を参照する。
教育、治療と遺伝カウンセリングのための包括的出血性疾患プログラムに紹介することはVWD患者に有益である。2つの主な治療法として、デスモプレッシン(1-デアミノ-8-Dアルギニンバソプレッシン[DDAVP])及びVWFと第Ⅷ因子を含む凝固因子濃縮物がある。VWD患者は出血が重度な場合に迅速な治療を受ける必要がある。
デスモプレッシン
ほとんどの1型VWD患者及び一部の2型VWD患者は、デスモプレッシンの静脈内注射もしくは皮下注射に反応する[Castaman et al 2008,Federici 2008,Leissinger et al 2014]。この治療法によって、貯蔵されていたVWFの放出が促進され、VWF値が34倍に上昇する。デスモプレッシンの経鼻投与もできる。
VWDと診断された後は、VWF反応を評価するためのデスモプレッシン負荷試験を行うのが望ましい。
デスモプレッシンは、急性の出血症状もしくは手術を対応するための治療選択肢である。
デスモプレッシンは、1型VWD女性患者の出産時の出血に有効であり、さらに一部の2型VWD女性患者の妊娠時にも投与している[Castaman et al 2010b]((妊娠管理の項を参照)。
デスモプレッシン耐性患者もしくはVWF応答性が悪い患者に対しては、凝固因子濃縮物投与が必要である。
デスモプレッシンは動脈系疾患を持つ患者や70歳以上の患者では禁忌であり、これらの患者にはVWF及び第Ⅷ因子濃縮物投与が必要である。
注意:デスモプレッシンは低ナトリウム血症(発作や昏睡が生じる)を引き起こす可能性がある。低ナトリウム血症のリスクを最小限にするために、デスモプレッシン投与後24時間は水分摂取を制限する必要がある。
濃縮VWF/第Ⅷ凝固因子製剤の静脈内注入
デスモプレッシンに応答しない患者(例えばVWF欠損が十分に治療されていない患者)およびデスモプレッシンが禁忌の患者([タイプ別VWDの治療]の項を参照)に対しては、VWF及び第Ⅷ因子を含む血漿由来の不活性濃縮凝固因子製剤を静脈内注入することによって、出血症状の予防もしくはコントロールができる[Federici 2007]。この濃縮製剤は多くのドナーによる献血から作られる。潜在的病原体はウイルス不活性化処理によって除去されている。
間接治療
VWFレベルを直接上昇させる治療に加えて、VWDの患者に対して、次の間接的止血治療もしばしば有効となる。
- 線維素溶解阻害剤(例えば出血症状の治療もしくは予防に用いられるトラネキサム酸);
- ホルモン療法(例えば月経過多の治療に用いられる複合経口避妊薬)。
タイプ別VWDの治療
1型VWD デスモプレッシンやVWF/第Ⅷ因子を含む濃縮凝固因子製剤のような直接VWF値を上昇させる治療は通常、大きな外傷や外科手術時の重度な出血の治療もしくは予防に対してのみ必要となる。
線維素溶解阻害剤やホルモン療法のような間接的治療はしばしば効果的である。
2A型VWD 濃縮凝固因子製剤を用いた治療は、通常外科手術中におこるような重度な出血の治療もしくは予防に対してのみ必要となる。
患者のデスモプレッシンに対する応答性は様々であるため、治療目的で投与する前に確認する必要がある。
間接的治療は有効である場合がある。
2B型VWD 濃縮凝固因子製剤は通常重度な出血の治療もしくは手術の際に必要となる。
血小板減少症を悪化させることがあるため、デスモプレッシンを用いた治療は慎重に行うべきである。但し、特定な病原性変異による軽度もしくは非定型的2B型VWDの患者において、デスモプレッシンへの暴露による血小板減少症は生じない[Federici et al 2009]。
間接的治療(例えば、線維素溶解阻害剤)は有効である場合がある。
2M型VWD 一般的に、デスモプレッシンに対する応答性は低いので、濃縮VWF/第Ⅷ因子製剤が治療の選択肢となる。
2N型VWD デスモプレッシンは軽度な出血に使用されるが、第Ⅷ因子レベルは急激に低下することがある(第Ⅷ因子がVWFに保護されていないので)ので、外科手術中は濃縮VWF/第Ⅷ因子製剤が必要となる。
3型VWD 通常濃縮VWF/第Ⅷ因子製剤を投与する必要がある[Franchini et al 2007]。3型VWDに対し、デスモプレッシンは有効ではない。間接的治療が効く場合がある。
小児科的問題
VWDの幼児や子供のケアに対して以下のような特別な注意が必要である。
- 男児に対する割礼は、小児血液専門医による診察の後に行うべきである。
- デスモプレッシンは注意して使用する必要がある。2歳未満の幼児は、水分摂取制限に対する潜在的な問題を伴う(水中毒を惹起しやすい。訳者注)ので特に注意が必要である。
- 新生児期はVWF値が比較的高いので、軽症VWDの新生児に対する表現型の検査は、幼児期後期まで延期すべきである。
一次病変の予防
3型VWD患者に対して、筋骨格出血およびそれに続発する関節損傷を予防するため濃縮VWF/第Ⅷ因子製剤の予防的投与がしばしば行われる。
二次病変の予防
デスモプレッシンは注意して使用する必要がある。2歳未満の幼児は、水分摂取制限に対する潜在的な問題を伴うので特に注意が必要である。
VWD患者にはA型およびB型肝炎の予防接種を行うべきである[Nichols et al 2008, Castaman et al 2013]。
サーベイランス
軽度なVWD患者が、出血性疾患のマネジメントに経験のある医療機関で経過観察を受けることは有益である。
3型VWD患者は、経験のある医療機関で経過観察を受けるべきである。また理学療法士による関節可動性の評価を定期的に行う必要がある。
避けるべき薬剤や環境
外傷、特に頭部外傷に高いリスクを伴う活動は避けるべきである。
出血症状を悪化させるので、血小板機能に影響を及ぼす薬剤(アセチルサリチル酸、クロピドグレル、非ステロイド系抗炎症薬)の使用は避けるべきである。
男児の割礼は小児血液専門医との相談の後に行わなければならない。
リスクのある血縁者の検査
家系内に病原性変異が同定された場合、リスクのある血縁者は速やかに遺伝学的検査をすることで、早期診断及び必要な治療を受けることができる[Keeney et al 2008]。
遺伝カウンセリングを目的として行われるリスクのある血縁者の検査に関する問題は、[遺伝カウンセリング]の項を参照。
妊娠中のマネジメント
VWF値は妊娠第三半期にピークに達し、妊娠期間中を通じて上昇する。それにもかかわらず、VWD妊婦は出血性合併症のリスクが増加するため、出血性疾患の周産期管理に経験のある医療機関でのケアが必要である[James & Jamison 2007, Varughese & Cohen 2007, James et al 2009]。
VWFおよび第Ⅷ因子の基準値が30IU/dL以上の女性は、分娩までの期間に正常値になることが多い。しかし、基準値が20IU/dL未満もしくはVWF:RCo/VWF:Ag比が0.6以下の女性において、代替療法が必要となることが多い[Castaman et al 2013]。
分娩は産科的兆候に基づいて行わるべきであるが、使用器具は最小限にすべきである[Demers et al 2005]。
遅延性、二次性の産後出血は問題となることがある。VWF値を分娩後速やかに妊娠前に戻る[Castaman et al 2013]。
研究中の治療法
現在臨床試験中の遺伝子組み換えVWFは、近いうちに利用可能となる見込みである。遺伝子組み換え第Ⅷ因子や第Ⅸ因子のように、遺伝子組み換えVWFが血漿由来VWFに代わって広く使われるようになると考えられる[Turacek et al 2010, Mannucci et al 2013]。
さまざまな疾患についての臨床研究の情報は、ClinicalTrials.govで検索できる。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ほとんどの1型、ほとんどの2A型、2B型、2M型は常染色体優性の遺伝形式をとる。
2N型、3型、1型の一部と2A型の一部は常染色体劣性の遺伝形式をとる。 片親性ダイソミーによる3型VWDが1例報告されている。2つの病原性変異アレルは母親ダイソミーによって一緒に受け継がれた[Boisseau et al 2011]。
患者家族のリスク:常染色体優性遺伝
発端者の両親
- 常染色体優性VWDと診断された患者のほとんどは、罹患している親を持つ。
- 常染色体優性VWDの発端者は、新規突然変異によるものが考えられる。新規突然変異の頻度は分かっていない。
- 発端者で見つかった常染色体優性VWDを引き起こす病原性変異が、両親のDNAで見つからなかった場合は、両親のいずれかの生殖細胞モザイクもしくは発端者の新規突然変異、2つの可能性が考えられる。しかしどちらの可能性についても十分に調査されていないので、相対的な発症率について言及できない。
- 常染色体優性VWDの発端者において、血縁者にVWD患者がいなかったこと、発端者の両親は症状が確認される前に他界したこと、あるいは罹患している親の出血症状が遅れたことによって、家族歴を認められなかった場合がある。しかし、発端者の両親に適切な検査を行わない限り(例えば、VWD止血因子検査および/または発端者の病原性変異が同定されていた場合の分子遺伝学的検査、)、家族歴が陰性であることは言い切れない。
発端者の同胞
- 発端者の同胞のリスクは、発端者の両親の遺伝学的状態による。
- 発端者の片親が罹患している場合、同胞のリスクは50%である。
- 発端者の親が臨床的に非罹患であっても、その親において浸透率が不十分である可能性が残るため、発端者の同胞は一般集団よりリスクは高い。
- 発端者の病原性変異が、どちらの親のDNAにも見つからなかった場合、同胞のリスクは低い。しかし生殖細胞系列モザイクの可能性が残るため、一般集団よりリスクは高い。
発端者の子
- 常染色体優性VWDの発端者の子は、50%の確率でVWF病原性変異を受け継ぐ。
発端者の他の家族
- 発端者の他の家族のリスクは、発端者の両親の遺伝学的状態による。片親の罹患が明らかな場合、その血縁者はリスクがある。
患者家族のリスク:常染色体劣性遺伝
発端者の両親
- 常染色体劣性VWDの患者の両親は、必然的にヘテロ接合型となる(すなわち一つのVWF病原性変異を持つ)。
- 3型VWDのヘテロ接合型(保因者)は通常無症状である。しかし、保因者の15~50%は軽度な出血症状が見られ、1型VWDと診断されることがある[Nichols et al 2008, Bowman et al 2013]。
- 2N型VWDのヘテロ接合型(保因者)は通常無症状である。しかし、ごく一部の保因者は軽度な出血症状が見られ、1型VWDと診断されることがある。
発端者の同胞
- 両親とも常染色体劣性VWF病原性変異保因者であれば、妊娠時、子どもは罹患者となる確率は25%、保因者となる確率は50%、罹患者でも保因者でもない確率は25%である。
- 罹患者の同胞が非罹患の場合、その同胞が保因者となる確率は2/3である。
- 両親の中に、一人だけが保因者である場合(片親ダイソミーの場合[Boisseau et al 2011])、発端者の同胞が罹患するリスクは分かっていないが、極めて低い(<1%)。しかし、発端者のすべての同胞は保因者となる確率は50%、罹患者でも保因者でもない確率は50%である。
- 3型VWDヘテロ接合型(保因者)は通常無症状である。しかし、保因者の15~50%は軽度な出血症状が見られ、1型VWDと診断されることがある[Nichols et al 2008, Bowman et al 2013]。
発端者の子
常染色体劣性VWDの患者の子は、必然的にVWF病原性変異のヘテロ接合型(保因者)となる。
発端者の他の家族
発端者の両親の同胞は50%の確率でVWF病原性変異の保因者となる。
保因者診断
家系内における病原性変異が同定されていれば、リスクのある血縁者の保因者診断が可能である。
遺伝カウンセリングに関連した問題
早期診断と早期治療を目的としたリスクのある血縁者に対する情報については[臨床的マネジメント]のリスクのある血縁者の内容を参照する。
明らかな新規突然変異を持つ家族が考慮すべき事項
常染色体優性遺伝形式の発端者の両親のいずれも病原性変異を持たず、さらに本症に罹患している臨床所見がない場合、発端者は新規突然変異の可能性が高い。しかしながら、親が生物学上の父もしくは母でない場合(例えば生殖補助医療が行われている場合)や未公開な養子縁組など、非医学的な可能性も検討する必要がある。
家族計画
- 遺伝学的リスクの決定、保因者状態の説明、出生前診断についての議論は妊娠前に行うべきである。
- 罹患者、保因者もしくはリスクのある若い成人に遺伝カウンセリング(子の潜在的遺伝学的リスクおよび生殖に関わる選択肢についての議論を含めて)を行うのが望ましい。
DNAバンク
検査の方法、遺伝子や変異、疾患の理解が将来向上する可能性は十分に考えられる。従って将来のために、罹患者のDNA(通常は白血球から抽出したもの)の保存を十分に考慮すべきである。現在行われている検査の感度が100%でない場合は、特にDNAバンキングを考慮すべきである。DNAバンキングのサービスを行っている機関を参照。
出生前診断
家系内におけるVWF病原性変異が同定されていれば、リスクのある妊娠(通常は3型VWD)に対し、出生前検査もしくは着床前遺伝学的診断は選択肢となる。
治療法のある疾患において、早期診断よりむしろ妊娠中絶を目的とした出生前診断は問題である。ほとんどの施設では出生前診断の選択を両親に委ねているが、これには議論の余地がある。
(訳注:日本国内ではVWDにおける出生前診断および着床前診断は実施していない。)
リソース
- フォンウイルブランド病 小児慢性特性疾病情報センター
- ヘモフィリアねっと 一般社団法人 ヘモフィリア友の会全国ネットワーク・サイト
http://hemophilia-japan.org/contents/knowledge/2-1-1/2-1-1-1.html
- ヘモフィリアステーション
分子遺伝学
Table A
フォンウィルブランド病:遺伝子とデータベース
| 遺伝子 | 染色体座位 | タンパク | 特異的座位 | GMD |
|---|---|---|---|---|
| VWF | 12q13.31 | フォンウィルブランド因子 | von Willebrand Factor Database ISTH-SSC VWF Online Database - VWF |
VWF |
Table B
OMIMにおけるフォンウィルブランド病関連情報
| フォンウィルブランド病1型、VWD1 | |
| 277480 | フォンウィルブランド病3型、VWD3 |
| 613160 | フォンウィルブランド因子、VWF |
| 613554 | フォンウィルブランド病2型、VWD2 |
遺伝子構造 VWFは52個のエクソンを持ち、長さが178kbの遺伝子である。VWF遺伝子産物は8.8kbのmRNAと2813個のアミノ酸からなるタンパクである[Sadler 1998]。遺伝子およびタンパクに関する詳細な情報はTable A、遺伝子を参照する。
正常アレル変異 正常アレル変異は極めて一般的である。最近、エクソンおよびエクソン/イントロンの隣接領域において500以上の正常変異は報告されており、正常アミノ酸置換は80残基にあると予測されている(Table 4)。北ヨーロッパ起源でない集団を検索したところ、更に多くの正常アレル変異は同定された[Bellissimo et al 2012,Johnsen et al 2013,Zhou et al 2014]。ある研究では、VWF病原性変異のスクリーニング検査を受けた対象者において、平均17個のヘテロ接合型正常変異は同定された[Hashemi Soteh et al 2007]。VWF遺伝子における多くの正常変異に加え、遺伝子サイズの大きさおよび部分的偽遺伝子VWFP(エクソン23-34)の存在によって、全遺伝子配列解析とデータ解釈が困難となる。
1型VWDにおいて、約10%の罹患者は1つ以上の配列変異が見られた。これらの配列変異はcis型(同じアレル)とtrans型(別々のアレル)の両方が存在する[Cumming et al 2006,Goodeve et al 2007,James et al 2007a]。これによって、病原性変異と正常アレル変異の鑑別が更に困難となった。同様に、プロモーター領域における配列変異も確認されており、ある1型VWD家系において、病原性変異である13bpの欠失が同定された[Othman et al 2010]。
連鎖解析に有用な正常アレル変異はTable 4で示している。中には、プロモーター領域とイントロン40における短い縦列反復配列(rs41402545とrs36115023)[Vidal et al 2005]、および頻度の高い一塩基変異を含む。
Table 4
抜粋されたVWF正常アレル変異
| DNA塩基配列の変化 | タンパク・ アミノ酸の変化 |
VWFエクソン /イントロン |
参考SNP 番号 |
制限領域/ 多型の種類 | 参考配列 |
|---|---|---|---|---|---|
| c.1451A>G1 | p.His484Arg | Exon 13 | rs1800378 | Rsa I | NM_000552.3 NP_000543.2 |
| c.1946-19_1946-17dupCTT1 | なし | Intron 15 | rs10622288 | 3-bp挿入/欠失 | |
| c.2365A>G1 | p.Thr789Ala | Exon 18 | rs1063856 | Rsa I | |
| c.2555A>G | p.Gln852Arg | Exon 20 | rs216321 | Nla IV | |
| c.4141A>G1 | p.Thr1381Ala | Exon 28 | rs216311 | Hph I | |
| c.4414G>C | p.Asp1472His | Exon 28 | rs1800383 | RleA I | |
| c.4641C>T1 | p.Thr1547Thr | Exon 28 | rs216310 | BstE II | |
| c.6187C>T | p.Pro2063Ser | Exon 36 | NA | ||
| c.6977-542_6977-541ins24 | なし | Intron 40 | rs36115023 | 欠失/挿入多型 | |
| c.6977-715_6977-714ins16 | なし | Intron 40 | rs41402545 | 欠失/挿入多型 | |
| c.8113G>A | p.Gly2705Arg | Exon 49 | rs7962217 |
上記は一般的な非同義変異のごく一部である。
- 正常変異はいくつかの民族集団もしくは/および感度の良い制限酵素切断部位に影響を与えるため、連鎖解析には有用である。
病的アレル変異 VWDのほとんどは1塩基置換(Table 5, Figure 1)が原因である[James & Lillicrap 2006]。質的異常(2型VWD)はVWFタンパク機能に重要な領域におけるミスセンス変異が原因である。1型VWDに見られる部分的な量的異常のほとんどはミスセンス変異によって生じる。3型VWDに見られる重度な量的異常のほとんどは、null-allelesを引き起こすホモ接合もしくは複合型ホモ接合が原因である。しかし中には、ごく一部ミスセンス変異によるものもある。病原性変異はISTH-SSC VWF Databaseに登録されている。
Table 5
VWF病原性変異の抜粋
| VWDのタイプ1 | DNA塩基の変化 | タンパク・アミノ酸の変化 | VWFエクソン | 参考配列 |
| 1 | c.3614G>A | p.Arg1205His | 27 | NM_000552.3 NP_000543.2 |
| 1 | c.4751A>G | p.Tyr1584Cys | 28 | |
| 2A | c.4517C>T | p.Ser1506Leu | 28 | |
| 2A | c.4789C>T | p.Arg1597Trp | 28 | |
| 2B | c.3797C>T | p.Pro1266Leu | 28 | |
| 2B | c.3916C>T | p.Arg1306Trp | 28 | |
| 2B | c.3946G>A | p.Val1316Met | 28 | |
| 2B | c.4022G>A | p.Arg1341Gln | 28 | |
| 2M | c.3835G>A | p.Val1279Ile | 28 | |
| 2M | c.4273A>T | p.Ile1425Phe | 28 | |
| 2N | c.2372C>T | p.Thr791Met | 18 | |
| 2N | c.2446C>T | p.Arg816Trp | 19 | |
| 2N | c.2561G>A | p.Arg854Gln | 20 | |
| 3 | c.2435delC | p.Pro812ArgfsTer31 | 18 | |
| 3 | c.4975C>T | p.Arg1659Ter | 28 | |
| 3 | c.7603C>T | p.Arg2535Ter | 45 |
変異分類に関する注意:上記に記載されている変異は作者が作成したもので、GeneReviewsはこれらの変異の分類を単独な検証を行わなかった。
命名法に関する注意:GeneReviewsはHuman Genome Variation Society (www?.hgvs.org)の標準命名規則に従う。命名に関する解釈はQuick Referenceを参照する。
- VWDの各型に同定されている頻度の最も高い変異を示している。各アレル変異および頻度に関する更なる情報はISTH-SSC VWF Databaseを参照する。
正常遺伝子産物 2813個のアミノ酸からなるVWFタンパクは、シグナルペプチド(22個アミノ酸)、プロペプチド(741個アミノ酸)と成熟タンパク(2050個アミノ酸)を含む。最近、VWFタンパクのドメイン構造は修正され、Figure 1で示している[Zhou et al 2012,Valentijn & Eikenboom 2013]。タンパク合成中、ジスルフィド結合(S-S)によるtail-to-tailの二量体はCKドメインを介して形成される。その続きにhead-to-head多量体が付加される。上記のプロセスを触媒するジスルフィド異性化酵素サイトはプロペプチドに存在する。VWF合成は内皮細胞と、血小板の前駆体である巨核球、2つの部位で行われる。内皮細胞と血小板から分泌されたVWFには、長さが最大40個のサブユニット(二量体)多量体が含まれる。多量体産生時に、プロペプチドは763と764番目のアミノ酸の間にフリンによって切断される。これらのプロペプチド(VWFpp)はVWFと一緒に血漿中に分泌される。VWFppと成熟VWF(VWF:Ag)の割合を用いて、成熟VWFの相対的半減期を推定できる[Haberichter et al 2008]。この割合から、病理学的メカニズムに関する情報を得られる[Eikenboom et al 2013]。
高分子量VWFの血栓形成性を抑えるために、分泌直後、1605と1606番目のアミノ酸の間にADAMTS13(ジスインテグリンと1型トロンボスポンジンモチーフをもつメタプロテアーゼ)によって切断される。この多量体タンパク分解は、各主要多量体バンドに隣接するサテライトバンドのような特徴的な「トリプレット」パターンを生成し、多量体解析ゲルで観察できる。バンドパターンの異常は、VWDのサブタイプの決定に手掛かりとなる[Schneppenheim & Budde 2011]。
VWFは主に2つの機能がある。(1)コラーゲンを血管損傷部位の内皮下層に結合し、血小板集積と血栓形成による血管修復を開始させる。(2)第Ⅷ因子を未熟なプロテアーゼによる分解から守り、フィブリン生成が必要な部位までに運ぶ。
異常遺伝子産物 異常VWFは病原性変異のタイプによって異なる。タンパクと塩基配列の両方における分子学的変化によって、異なるVWDが生じる。
- 1型VWD ほとんどはミスセンス変異であるが、VWFに影響を及ぼすメカニズムは異なる。1型VWDに対し、分子遺伝学的検査を行うのは最近のことなので、多くの症例において病理学的メカニズムはまだ確認されていない。
- ほとんどのミスセンス変異はD3とA1ドメイン[Haberichter et al 2008, Millar et al 2008, Eikenboom et al 2013]にある。これらのミスセンス変異によって、VWFの血漿中の滞留時間が数倍に減少する。"Vicenza"変異とも呼ばれるp.Arg1205Hisはその中に最も代表的で且つ一般的である。このような病原性変異は「VWFにおける血栓および止血科学と標準化委員会(ISTH SSC on VWF)」が承認したVWD分類ではないが[Sadler et al 2006]、1C型(type 1 clearance)とも呼ばれる[Haberichter et al 2006]。
- 細胞内貯留は病理学的に1型VWDに共通するメカニズムである[Eikenboom et al 2009,Eikenboom et al 2013]。
- 割合が小さいが、ヘテロ接合型null-alleleによるVWF発現が減少し、ハプロ不全(haploinsufficiency)は認められている。
- 2A型VWD ミスセンス変異が原因で、いくつかの機構の共同作用によって、高分子量多量体、時には中分子量多量体の欠失が見られる。これらの機構には、(1)異常二量体の蓄積、(2)異常多量体の蓄積、(3)ADAMTS13がコードするVWF切断プロテアーゼへの感受性の上昇[Hassenpflug et al 2006]、および(4)細胞内貯留[Schneppenheim et al 2010]がある。その結果、高分子量VWFが欠損し、GpIbαとの結合部位が減少することによって、有効な血小板血餅形成が減少する。
- 2B型VWD ミスセンス変異によって、VWFが血小板糖タンパクGpIbαとの結合能が上昇する。その結果、通常内皮下損傷後にVWFがコラーゲンと結合するための構造変化がなくても、VWFとGpIbαの結合(血小板-VWF複合体)が連続的に起きる。血小板-VWF複合体は循環系から除去され、血小板減少が生じる。また、高分子量多量体は優先的に血小板と結合するため、その減少はより顕著である。血小板と結合したVWFはさらにVWF切断プロテアーゼ(ADAMTS13がコードする)への感受性が高まることも、高分子量多量体の著減の一因である。循環系からVWFの過剰な除去は、表現型に大きな影響を与える[Casonato et al 2010]。病原性変異p.Pro1266Leuおよびp.Arg1379Cysは、GpIbα結合能の上昇だけに関与するが、低血小板血症や高分子量多量体の欠損に関与しない[Federici et al 2009, Casonato et al 2010]。
- 2M型VWD A1ドメイン(Figure 1)にあるミスセンス変異が原因で、VWFタンパクの立体構造が変化し、2A型のように高分子量多量体欠損がなくても、A1ドメインがGpIbαとの結合ができなくなる[James et al 2007b]。それに対し、A3ドメインにあるミスセンス変異は、VWFがGpIbαとの親和性が低下し、内皮下コラーゲンとの親和性が低下する。
- 2N型VWD 第Ⅷ因子の結合部位における重要なアミノ酸置換もしくは立体構造変化によって、VWFが第Ⅷ因子との親和性が低下する。これは、VWF-第Ⅷ因子結合に非直接的な影響を与える。
- 3型VWD 両方の対立遺伝子に、VWFの分泌障害を来す病原性変異(nullまたはミスセンス)が生じている。3型VWD患者のほとんどは2本のnull-allelesを持ち、その結果、VWFの産生量が著減する。約20%の対立遺伝子において、D1またはA1-A2ドメイン(Figure 1)に影響するミスセンス変異がある。これらのミスセンス変異の一部はVWF多量体化を障害し、VWFの細胞内貯留および血漿への分泌閉止を来たす
更新履歴
- Gene Review著者: Anne C Goodeve, PhD
日本語訳者: 坂下建人(旭川医科大学医学科学生),蒔田芳男(旭川医科大学 教育センター)
Gene Review 最終更新日: 2009.6.4. 日本語訳最終更新日:2010.7.13 -
Gene Review著者: Anne C Goodeve, PhD
日本語訳者:江田肖(瀬戸病院遺伝診療科),櫻井晃洋(札幌医科大学附属病院遺伝子診療室)
Gene Review 最終更新日: 2014.7.24. 日本語訳最終更新日: 2016.11.16. (n present)