網膜芽細胞腫
(Retinoblastoma)
GeneReview 著者:Dietmar R Lohmann, MD; Brenda L Gallie, MD.日本語訳者: 島田咲(京都大学大学院医学研究科),山田崇弘(京都大学医学部附属病院遺伝子診療部)
GeneReview最終更新日: 2018.11.21 日本語訳最終更新日: 2016.9.7
要約
疾患の特徴
網膜芽細胞腫(Rb)は通常5歳までの小児に発症する、発達中の網膜におこる悪性腫瘍である。RbはRB1遺伝子の両コピーに発癌の素因となる変異が生じた細胞から発症する。Rbは単発性の場合もあるし、多発性の場合もある。Rb患児の約60%は片側性で診断時の平均月齢は24か月、約40%は両側性で診断時の平均月齢は15か月である。遺伝性の網膜芽細胞腫とはRb感受性が常染色体優性で遺伝するものである。遺伝性Rbの患者においては、眼以外の腫瘍を発症するリスクも増大する。
診断・検査
網膜芽細胞腫の臨床診断は、間接的検眼鏡を用いた眼底検査によってなされる。画像診断は臨床診断を支持し、病期を評価する目的で行われる。遺伝性の網膜芽細胞腫は、Rbまたはレチノーマの発端者で家系内にRbの既往歴がある場合や、RB1遺伝子における生殖細胞系列の変異を同定した場合に診断される。
臨床的マネジメント
症状の治療:
Rbおよび眼以外の腫瘍の早期診断と治療によって罹病の減少と寿命の延長が可能である;
治療にあたっては、眼科学、小児腫瘍学、病理学、放射線腫瘍学といった複数の領域の専門家チームによるのが最善である。治療の選択肢は、腫瘍のステージ、病巣の数(単一または片側性、 多巣性または両側性か)、眼球中の腫瘍の位置やサイズ、硝子体播種の有無、視力を残存できる可能性の有無、眼以外への侵襲の程度、その他あらゆる可能性を加味して決定される。治療の選択肢には、眼球摘出術、凍結療法、レーザー、レーザーまたは凍結療法との組み合わせで、またはその後に行われる、動脈内化学療法を含めた 眼球に限定もしくは全身にわたる化学療法、強膜上のプラークを利用した放射線療法、そして最後の手段としての外部からの放射線療法がある。
二次発症の予防:
晩発性の二次がんを発症するリスクを最小限にとどめるために、遺伝性 Rb の患者に対する放射線照射(X線、CTスキャン、外部照射を含めて)は可能な限り避けるべきである。
サーベイランス:
RB1遺伝子の生殖細胞系列の病的変異が同定されている患児には 、1歳までは3から4週間毎の眼の検査、3歳まではそれよりはやや間隔を広げた頻度での検査が推奨される。協力的な患児の臨床検査は7歳までは3から6か月毎に、その後は毎年から隔年での検査が生涯続けられる。片側性 RbでRB1遺伝子の生殖細胞系列における病的変異が同定されていない患児は低レベルのモザイクである可能性があり、超音波検査を含めた定期的な眼の検査を受けることが推奨される。 網膜細胞腫患者は網膜の検査を1から2年ごとに受けるべきである。網膜芽細胞腫の罹患者は肉腫や他の癌のリスクが高いので、眼以外におこる二次腫瘍の発見のためには、担当医や親は骨の痛みや腫れがあるという訴えがあったら、すみやかに対処するべきである。しかし、効果的なスクリーニング・プロトコールはまだ確立されていない。
避けるべき環境:
DNA損傷因子(放射線、たばこ、紫外線)への暴露を最小限にとどめることによって、遺伝性の網膜芽細胞腫の罹患者の発がんリスクを減らすことが出来る。
リスクのある血縁者 の検査:
リスクはあるが症状のない子に対する早期の分子遺伝学的解析によって、病的変異を受け継いでいない家族に対するコストのかかるスクリーニングを削減することが出来る。
遺伝カウンセリング
遺伝性の網膜芽細胞腫は常染色体優性遺伝形式で受け継がれる。遺伝性 網膜芽細胞腫の患者は、親から受け継いだかまたは新生突然変異によって起こったRB1遺伝子の病的変異を生殖細胞系列にヘテロで持つ。患者の子 は50%の確率で病的変異を受け継ぐ。リスクのある妊娠における出生前検査は家系におけるRB1遺伝子の病的変異が同定されている場合に限り可能である。
診断
網膜芽細胞腫に罹患した子 とその家族に対する診断と治療のガイドラインが出版されている[Canadian Retinoblastoma Society 2009]。
疑い所見
網膜芽細胞腫は以下の所見によって疑われる。
- 白色瞳孔
- 斜視
- 眼の外見的な 変化
- 視力の低下
遺伝性の網膜芽細胞腫は以下に示す所見と家族歴によって疑われる
- 片側性(単巣性、多巣性を問わず)と両側性 を含め 、Rbの診断を受けた患者
- 網膜細胞腫患者
- 網膜芽細胞腫の家族歴がある 者
臨床診断
発端者における網膜芽細胞腫の診断は、眼科医か 眼鏡医(optometrist)による瞳孔の完全散大後の検査によってなされる。診断の確認、病気の進行度の確認は麻酔下の検査で行われる。眼の画像診断は診断の確定に役立つ。病理診断は必要ない。注:生検によって腫瘍が眼以外に拡大することがあり、患者の生命を脅かす可能性がある。
遺伝性の網膜芽細胞腫は、Rbの発端者 または網膜細胞腫 と家族歴 がある場合に診断される。Rb患者の大部分は家族歴を持たない。そのため、分子遺伝学的解析によるRB1遺伝子の病的変異の同定(Table 1参照)は、発端者のRbが遺伝性かどうかを決定し、Rbのリスクのある親族において早期に診断とスクリーニングを行うために必要不可欠である。
遺伝性網膜芽細胞腫の診断のための分子生物学的検査には、単一遺伝子の検査とマルチジーンパネルを用いた検査とがある。
単一遺伝子の検査
- 両側性、片側性で家族性、片側性で多巣性Rbの患者
RB1遺伝子のシークエンス解析と遺伝子特異的な欠失/重複解析を、末梢血DNAを用いて行う。注:メチル化CpGジヌクレオチドにおける病的変異の解析を行う施設もある。
- 片側性で単巣性のRbで家族歴がない患者
- 腫瘍組織が得られない場合は、末梢血DNAを用いてRB1遺伝子のシークエンス解析と遺伝子特異的な欠失/重複解析を行う。
- 腫瘍組織が入手可能な場合は、RB1遺伝子のシークエンス解析と遺伝子特異的な欠失/重複解析を腫瘍DNAを用いて行う。病的変異が同定された場合は、血液においても当該変異の有無を検査する。病的変異が同定されなかった場合、RB1遺伝子のプロモーター領域における 高メチ ル化によってRB1遺伝子のエピジェネティックな不活性化がないか確認するために、RB1遺伝子プロモーターのCpGアイランドのメチル化解析を行う。プロモーターに 高メチル化が見られなかった場合、腫瘍DNAを用いて、MYCNの増幅がないか検査する、これは片側性の網膜芽細胞腫患者の約1.5%においてRB1遺伝子の病的変異が見られない場合のRbの原因となっている。
マルチジーンパネル
RB1遺伝子を含むマルチジーンパネルも選択肢の一つである。しかし、遺伝性の網膜芽細胞腫では遺伝子座異質性は知られていないことから、解析データの解釈はRB1遺伝子の変異による。
表1 遺伝性の網膜芽細胞腫において用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | 試料 | 生殖細胞系列に病的変異2を持つ 発端者における変異検出率 |
|---|---|---|---|
| RB1 | シークエンス解析3 | 生殖細胞系列 腫瘍 |
70-75% |
| 当該遺伝子の欠失/重複解析4 | 生殖細胞系列 腫瘍 |
8-16%5 | |
| 病的変異のターゲット解析 | 生殖細胞系列 腫瘍 |
25%6 | |
| メチル化解析 | 腫瘍 | 注7参照 | |
| MYCN | 当該遺伝子の欠失/重複解析4 | 腫瘍 | 注8参照 |
- 染色体位置と遺伝子名については表A参照
- 変異については分子遺伝学的検査の項目を参照
- シークエンス解析によって得られる結果は、benign(良性)、likely benign(おそらく良性)、 of uncertain significance(意義不明)、likely pathogenic(おそらく病的)、pathogenic(病的)がある。病的変異には微小欠失/重複、ミスセンス変異、ナンセンス変異、スプライスサイトの変異が含まれ、エクソン単位、遺伝子レベルでの欠失/重複は少ないのが特徴である。シークエンス解析の結果の解釈は原文のこの注釈部分にリンクがある。
- 一遺伝子をターゲットとした欠失/挿入解析によって遺伝子内の欠失重複を検出することがある。検出法には定量PCR、ロングPCR、MLPA法、単一のエクソンの欠質または重複の検出のためにデザインされたマイクロアレイ法がある。
- 腫瘍におけるヘテロ接合性の消失(LOH)の検査。末梢血または腫瘍由来DNAからのRB1遺伝子内または隣接領域における多型の競合的タイピングによって、変異アレルの重複を伴う(ホモ接合性)または伴わない、正常遺伝子の欠失(ヘミ接合性)が体細胞における病的変異を構成していることが判明する。
- CpG変換による早期の転写停止による病的変異が病的変異全体の25%を占める[Rushlow et al 2009]。
- RB1遺伝子プロモーター領域の高メチル化(遺伝子発現を抑制する)は散発性、片側性の網膜芽細胞腫の患者の腫瘍の10%から12%にみられる[Zeschnigk et al 2004]。このような患者では、腫瘍の発生にかかる2つのRB1アレルの不活化を証明するためにはプロモーター領域のメチル化状態の解析が必要である。
- 散発性の片側性Rb患児の約1.5%では、腫瘍組織の検査においてMYCN遺伝子の高レベルの増幅が見られるが、RB1遺伝子の不活化をもたらす病的変異は検出されない[Rushlow et al 2013]。
表2 家族歴と腫瘍の性質に基づく網膜芽細胞腫患者におけるRB1遺伝子の生殖細胞系列変異の有無の可能性
| 家族歴 | Rbの表現形 | RB1の生殖細胞系列変異の可能性 | ||
|---|---|---|---|---|
| 片側性 | 両側性 | |||
| 多発性 | 単発性 | |||
| あり注1 | + | 100% | ||
| + | 100% | |||
| + | 100% | |||
| なし注2 | + | ほぼ100%注3 | ||
| + | 14~95% | |||
| + | ~14% | |||
注1:あり=家族内に複数の患者(網膜芽細胞腫の10%)
注2:なし=家族内に一人のみの患者(網膜芽細胞腫の90%)
注3:RB1遺伝子の病的変異は、両側性を含む単独のケースの90-97%では分子遺伝学的検査で証明されるが、残りの5%は、転座、イントロン内部でのスプライス変異、低レベルのモザイクなどが含まれ、生殖細胞系列に存在する場合としない場合とがある。
注:
- 腫瘍細胞において見出された変異が非腫瘍細胞(体細胞DNA)にはなかった場合、患者がRB1遺伝子に生殖細胞系列の変異を持つ可能性は低い。
- シークエンス解析のような簡便な分子解析で20%程度の低い割合のモザイクであっても検出できるため、RB1遺伝子の病的変異が白血球細胞に検出されないということは、その患者が生殖細胞系列の変異を持つ可能性が低いということは出来るが、その可能性を完全に否定するものではない。
遺伝的関連疾患
このGenereviewsで述べた以外のRB1遺伝子の病的変異に関連する表現型は知られていない。
臨床像
自然経過
網膜芽細胞腫(Rb)
最も頻度の高い初発症状は白色瞳孔反射(leukocoria)である。斜視は次に頻度の高い初発症状であり、白色瞳孔に随伴したり白色瞳孔より早期に見られたりする[Abramson et al 2003]。まれな初発症状としては、緑内障、眼窩蜂巣炎、ぶどう膜炎、前房出血、硝子体出血がある。患児のほとんどは5歳までに診断される。非典型的な症状は年長の患児で見られることが多い。
Rbの発端者は以下に示す臨床像のうちどれかを有することが多い。
- 家族歴のない片側性Rb(発端者の60%)
- 家族歴のない多発性 Rb(発端者の30%)
- 家族歴のある片側性または両側性のRb(発端者の~10%)。家族歴があり、網膜の検査による臨床サーベイランスを実施している人では、腫瘍はほとんどの場合、生後1ヶ月の間に発見される。
- 13q14を含む染色体欠失。単発性 Rbの発端者の最大5%および多発性 Rbの発端者の7.5%は、13q14を含む染色体欠失を有する。このような染色体異常を伴う患者はしばしば発達遅滞や先天奇形を伴う[Mitter et al 2011, Castéra et al 2013]。
網膜芽細胞腫は、
- Unilateral(片側性) 片眼だけがRbに罹患している場合。罹患者の約60%は片側性のRbで平均診断年齢は24ヶ月である。片側性Rb腫瘍はたいてい単巣性である、つまり、単一の腫瘍のみが存在する。片方の眼に複数の腫瘍を生じる場合もある(片側性(unilateral) 多巣性(multifocal)retinoblastoma)。眼球内播種は多巣性の腫瘍増殖に類似している。片側性Rb患者のほとんどは家族歴がなく、腫瘍が大きいために、単一の腫瘍かどうか診断できない場合が多い。
- Bilateral(両側性) 両眼がRbに罹患している場合。罹患者の約40%は両側性のRbで平均診断年齢は15ヶ月である。両側性の腫瘍を持つ患児のほとんどは最初の診断時に両眼に腫瘍が発生している。両方の眼に両側性Rbを持つ患者は、複数の腫瘍を発症することがある。初診時に片側性Rbと診断された患児のなかには後に反対の眼に腫瘍が発生することもある。
- Trilateral 両側性(ごくまれに片側性)のRbに松果体腫瘍が合併したもの。
網膜細胞腫と関連眼病変
網膜瘢痕中で自発的な成長の停止を引き起こす良性の眼球の腫瘍( 網膜細胞腫と呼ばれる)。眼の石灰化、退縮は血管閉塞によるRbの自然退縮によるものである [Valverde et al 2002] 。
松果体腫瘍
松果体腫瘍は、松果体にある"網膜様組織"に発生する。Rbとともに松果体腫瘍を発生する例をtrilateral Rbと呼ぶ。松果体腫瘍はまれであるが、眼に発生するRbが通常治癒可能であるのに対して、通常致死的である[de Jong et al 2014]。
その他の腫瘍
他の特定の眼以外の腫瘍(正しくは2番目の原発 腫瘍second primary tumorsと呼ぶ)の発生リスクも上昇する。ほとんどの場合、骨肉腫、軟部組織肉腫(ほとんどは平滑筋肉腫、横紋筋肉腫)、または黒色腫である[Kleinerman et al 2007, Marees et al 2008, Kleinerman et al 2012]。これらの腫瘍は思春期か成人期に発症することが多い。2番目の原発 腫瘍の頻度は外部からの放射線照射の治療を受けた患児の50%以上にのぼる[Wong et al 1997]。高容量の照射治療を受なかった遺伝性の網膜芽細胞腫患者は生涯にわたり後発性のがんのリスクが高まる[Fletcher et al 2004, Kleinerman et al 2012, Dommering et al 2012b, Temming et al 2015]。遺伝子型と臨床型の関連
大多数の遺伝性網膜芽細胞腫 家系では、生殖細胞系列の遺伝子変異を持つ全ての 血縁者において両眼に複数の腫瘍が発生する。しかし、最初に罹患した患者が片側にしか腫瘍が発生しないことは珍しくはない。こうした家系のほとんどでは、フレームシフトやナンセンス変異によって生じたRB1の"null" アレルを受け継いでいる。ごく少数の例外を除いて、RB1遺伝子の"null" アレルは浸透率99%以上の完全浸透の形で受け継がれる[Lohmann et al 1996, Sippel et al 1998]。
表現型が軽くなることによって浸透率が一見低く見える家系(片側性Rbに多い)や不完全浸透(25%以下)の家系もあるが、全体の10%以下である。このように浸透率の低い家系は、変異RB1 アレルが明らかなインフレーム変異、ミスセンス変異、特定のスプライスサイトの変異、エクソン1の特定の indel変異、プロモーター領域の病的変異と関連しているといわれている。
第3のカテゴリーの家系では、どちらの親由来の病的変異かで、家系内で異なる浸透率を示す(片親起源効果;一方の親家系でのみ連鎖が見られる遺伝) [Klutz et al 2002]。
通常の染色体検査で検出される13q14の欠失は、同領域にあるRB1以外の遺伝子群の欠失も伴うが、これを有する患児は発達遅滞[Castéra et al 2013]と軽度~中等度の顔面奇形徴候を呈する。13q14のかなりの大きさの欠失では表現型は軽くなる。こうした欠失を有する患児の多くは片側性のRbを呈し、腫瘍を発症しない場合もある[Mitter et al 2011]。RB1遺伝子のセントロメア側に隣接したMED4遺伝子の 欠失によって、RB1遺伝子とMED4遺伝子の両方にまたがる大きな 欠失をもつ患者における症状の軽症化が説明される[Dehainault et al 2014]。
浸透率
遺伝型と臨床型の関連の項参照
病名
網膜芽細胞腫の古くからの別名はGlioma retinaeである.
頻度
Rbの頻度は1人/15,000~20,000出生児と推定されている[Moll et al 1997, Seregard et al 2004]。
鑑別診断
本稿で扱われる疾患に対する遺伝学的検査の実施可能性に関する最新情報は,GeneTests Laboratory Directoryを参照のこと.―編集者注.
小児期に見られる以下のような眼疾患は臨床的に網膜芽細胞腫と類似している。
- 持続性 原発性硝子体過形成やCoats病(OMIM 300216)を含む孤発性の先天性疾患
- 結節性硬化症、ノリエー病、色素失調症、家族性滲出性硝子体網膜症(Autosomal Dominant Familial Exudative Vitreoretinopathy参照)、von Hippel-Lindau病などの遺伝性疾患。
- 眼におけるイヌ回虫の外寄生。
臨床的マネジメント
網膜芽細胞腫(Rb)治療のガイドラインが策定されている。(Canadian Retinoblastoma Society 2009)
最初の確定診断後の評価
Rbと診断された患者の病気の程度を確定するために以下の評価が推奨されている。
- 治療計画をたてる前に眼内、外の腫瘍の程度を見極めるべきである。罹患した眼は、病変の程度、腫瘍が眼球外に拡がる可能性によって分類される。家族歴がない場合、罹患した眼には大きな腫瘍があり、瞳孔を介して直接白色瞳孔反射(white pupillary reflex)として観察されることが多い。腫瘍の程度は、特に腫瘍と視神経との関係に主眼をおいて、麻酔下で超音波やMRIによって診断される。生殖細胞系列のRB1病的変異を持つ患児への照射の危険性から、CT スキャンは禁忌である。頭部MRIは三側性腫瘍を意味する松果体腫瘍の検出の上で有用である。
- 眼外症状のリスクを伴う大きな腫瘍の診断の際や、摘出した眼球が視神経への浸潤を伴う場合や脈絡叢への浸潤が発見された場合においては、骨髄穿刺と脳脊髄液の検査が行われる場合もある。
- Rbが眼外に拡がった場合には、患児への最適な治療のため癌のステージを再評価すべきである。
- 稀に、Rbの家族歴があり、患児が斜視か視力障害を呈している時には、腫瘍は小さく、麻酔下の検査で発見されることもある。
- 臨床遺伝専門医、遺伝カウンセラーへの紹介が推奨される。
病変の治療
治療の目標は第一に生命、そして視力 を 守ることである。最適な治療は多岐にわたっているため、Rbの治療経験が豊富な眼科、小児腫瘍、病理、放射線などの専門医がチームで診療する。
治療法の選択には、腫瘍の程度(眼内、眼外)、腫瘍の病期に加えて、腫瘍の数(単発性か、片側性で多 巣性か、両側性か)、眼内腫瘍の位置や大きさ、硝子体播種の有無、視力を残す可能性、眼外への拡散と程度、あらゆる情報を加味して行う。
治療の選択肢には、眼球摘出術、冷凍凝固療法、レーザー、レーザーまたは凍結療法との組み合わせで、またはその後に行われる、動脈内化学療法を含めた眼球に限定もしくは全身にわたる化学療法、 放射線療法(アイソトープ強膜上癒着照射)、そして最後の手段としての外部からの放射線療法がある。
二次発症の予防
晩発性の二次がんを発症する リスクを最小限にとどめるために、遺伝性 Rb の患者に対する放射線照射(X線、CTスキャン、外部照射を含めて)は可能な限り避けるべきである。これらの検査が絶対的に必要な場合にのみ行われるべきである。
サーベイランス
Rbを発症した患者またはリスクのある患者への調査に関する情報は網膜芽細胞腫治療ガイドラインに記載されている。
最初の診断後の Rbの検出
Rbの治療に成功してからも、定期検診を続け新しい眼内腫瘍の早期発見につとめる必要がある。
- 生殖細胞系列のRB1遺伝子の病的変異を持つ患児は、6か月までは麻酔下での3から4週間毎の眼の検査が、3歳まではそれよりはやや間隔を置いての検査が推奨される。クリニックでの検査は、協力的な患児では7歳までは3から6か月毎に、その後毎年から隔年での検査が生涯続けられる。
- 片側性のRbを持ち、生殖細胞系列でヘテロの病的変異が検出されない患児 は、低レベルのモザイクを持ち、もう一方の眼にも腫瘍を生じるリスクがある[Rushlow et al 2009, Temming et al 2013]。しかしそのリスクは小さいので、麻酔下の検査の代わりに定期的な超音波検査(シンプルで非侵襲的な検査)などに変更することも可能である。
- 網膜細胞腫(Rb関連の前癌性の網膜疾患)患者では、早期に病変を発見するために、1年から2年ごとに網膜の検査と画像検査を行う。
網膜芽細胞腫患者における2番目の眼外腫瘍の検出
肉腫、メラノーマ、その他の特異的ながんを含む二次がんのリスクが高いため、いかなるサインや症状に対しても迅速に対応することが推奨される。定期的な全身のMRI検査が、生殖細胞系列にヘテロのRB1遺伝子の病的変異を持つ患者における二次がんのスクリーニングに有効であるかについての検証が進行中である。
避けるべき環境
Fletcherらによると、DNA損傷因子(放射線、たばこ、紫外線)への暴露を最小限にとどめることによって、遺伝性のretinoblastoma保因者の発がんリスクを減らすことが出来る。こうした人においては、化学療法への暴露を最小限にすることが発がんリスクを低下させることは確かなようである 。
リスクのある親類への対応
米国臨床 腫瘍学会 (American Society of Clinical Oncologists: ASCO)は遺伝性の網膜芽細胞腫をグループ1(遺伝学的検査がリスクのある家族メンバーに対する標準的マネジメントの一部である遺伝性疾患)と判定している。明らかな 症状のない患者の 血縁者は、なるべく早期に、経験のある眼科医による検査を受け、Rbの早期発見につなげることが適当である。
以下の項目を実施する:
- 家系における病的変異が特定されている場合は、分子遺伝学的解析を行う、これによって病的変異を受け継いでいない 血縁者は多大なコストのかかるスクリーニング を省略することができる[Noorani et al 1996, Richter et al 2003]。
- 上述のように、出生後すぐにRbの治療に経験のある眼科医による眼の検査を行う。幼い、協力の得られない子どもには麻酔下での検査を行う。
リスクのある親族の検査の遺伝カウンセリングのための情報は、遺伝カウンセリングの項目を参照
調査中の治療法
広範囲にわたる病気における臨床検査に関する情報にはClinicalTrials.govサイトより入手できる。
この病気に関する臨床試行はない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
遺伝性の網膜芽細胞腫は常染色体優性遺伝である。
血縁者のリスク
患児の親
- Rbと診断された患児の親は罹患している場合がある。
- 遺伝性Rbの発端者は、RB1遺伝子の生殖細胞系列における新生突然変異によって発病することもある。大多数の遺伝性網膜芽細胞腫の患者はで家族歴がない場合は、新生突然変異によって罹患する。
- 発端者における病的変異が両親のどちらの白血球DNAからも検出されない場合、両親のどちらかが生殖細胞系列のモザイクであるか、発端者の生殖細胞系列における新生突然変異であるかの2つの可能性がある。モザイクの確率は約6%である。モザイクは、アレル特異的PCR [Rushlow et al 2009]か次世代シークエンサー[Chen et al 2014] のような高感度の方法で検出することができる。
- 明らかに新生突然変異の病的変異を持つ発端者の両親には分子遺伝学的検査が推奨される。RB1遺伝子における病的変異がわかっていない場合は発端者の両親に推奨される対応策は、Rb、 網膜細胞腫、Rb関連眼疾患に経験豊富な眼科医による診察である。
- 遺伝性Rbと診断された患者の家族歴は、家系内の疾病( 網膜細胞腫)の認識不足、低レベルのモザイク、浸透率の低下によって家族歴がないと判断されることがありうる。そのため、発端者の両親の適切な臨床検査 および/または、分子遺伝学的検査を行うことなしに家族歴がないと判断することはできない。孤発例とされる生殖細胞系列のヘテロのRB1遺伝子の病的変異をもつ患者の約10%で、発端者の両親のどちらかが病的変異を持っていた。ほとんどはモザイクか、ミスセンス変異のような浸透率の低いヘテロの変異を持つケースであった[Rushlow et al 2009]。
- 発端者がRB1遺伝子の病的変異のモザイクによってRbに罹患した場合、両親は病的変異を持たない。
注:親が最初に起きた病的変異を持つ場合、その親は体細胞性モザイクを持ち、まれに片側性のRbに罹患するかまたはRbには罹患しない。
患児の同胞
- 同胞のリスクは表現型と親の遺伝的状態による。
- 親がRB1遺伝子の生殖細胞変異を有していることが明らかであれば患児の同胞がRB1遺伝子変異を受け継ぐ確率は約50%となる。まれに「家族性で低浸透率のRb」の家系があり、その家系では病的変異を持つ同胞が腫瘍を発生する可能性は低くなる。
- 両親が臨床的に罹患していない場合、発端者の同胞のリスクは低い(1-2%、Table3参照)
- 臨床的に罹患していない両親を持つ発端者の同胞であっても、遺伝性Rbのリスクはなくはならない、なぜなら、両親が低浸透率の変異をもつ可能性があるからである。
- 発端者にみられるRB1遺伝子の病的変異が両親のどちらの白血球DNAからも検出されない場合、同胞のリスクは低いが生殖細胞系列のモザイクの可能性があるため、一般集団よりは高くなる。そのため、それぞれの同胞が発端者にみられるRB1遺伝子の病的変異の検査をすることが推奨される。
- 発端者が、白血球DNAのような非癌細胞において、腫瘍を発生しうるRB1遺伝子の変異を明らかにモザイクで持つ場合、病的変異は受精後に発生し、両親は生殖細胞系列にRB1遺伝子の病的変異を持たないと推定される。この場合同胞のリスクは高くならないため、発端者にみられるRB1遺伝子の病的変異の検査を正当化するものではない。
- 分子遺伝学的検査が実施できなかったり、有意な結果が得られない場合は、腫瘍の発生状況(単発性か多発性か)や家族歴に基づいた経験的リスクが用いられる(表3)。家族歴がない患児の同胞における低いが無視できないリスクは、おそらく1人の親が浸透率の低いRB1遺伝子の生殖細胞系列の変異を有していること、または1人の親が生殖細胞系列を含むRB1遺伝子変異の体細胞モザイクを有していること、を反映していると推測される。
- 一方の親が細胞学的検査で検出されうるレベルの13番染色体の転座や再構成を持つ場合、同胞は染色体のアンバランスを受け継ぐリスクを有する。
発端者の子
遺伝性Rb患者の子供は50%の確率でRB1遺伝子の病的変異を受け継ぐ。
- 患者が孤発性であるが両側性のRbを有していたら、その子どもがRB1遺伝子の生殖細胞系列の変異を有するリスクは約50%である。発端者のRB1遺伝子変異が判明している場合は子供の発症前検査が可能である。
- 患者が孤発性で片側性であるが多 巣性のRbを有していたら、その子どもの再発リスクは少ない[Sippel et al 1998, Rushlow et al 2009]。
- 単発性で単眼性でありまた家族歴がない患者の子どものリスクは、6%であり、このことは患者が低浸透率のRB1遺伝子の生殖細胞系列の変異を持っている、または患者が性腺を含む体細胞モザイクを持っている可能性を示している。家族性で低浸透率の網膜芽細胞腫の家系における発がんのリスクは、高浸透率のRB1遺伝子nullアレル家系における95%よりは低い。
- 発端者の白血球DNAにおいて、腫瘍細胞で検出されたRB1遺伝子の病的変異が検出されない場合、発端者が腫瘍細胞で検出された病的変異を生殖細胞系列のモザイクで持つ可能性は推定で1.2%ある。子供に生殖細胞系列の病的変異を受け継ぐリスクは0.6%である[Richter et al 2003]。子供の分子遺伝学的検査においては発端者の腫瘍で検出された2つの変異アレルの両方を検査するべきである。
- 発端者の白血球DNAで検出された病的変異のうち1つがモザイクである場合、生殖細胞系列にその変異を含む割合は不明である。すべての子供は白血球DNAで検出された病的変異の検査を受けるべきである。
表3 患者の同胞や子どもがRbを発症するかどうかについての経験的リスク(RB1生殖細胞系列の変異が見いだされていない場合)
| 患者の腫瘍発生状況 | 家族歴 | 患者の同胞 | 患者の子 | ||||
|---|---|---|---|---|---|---|---|
| 両側性 | 多発性 | 単発性 | |||||
| × | なし | 2%1 | 50% | ||||
| × | なし | 1-2%1 | 6-50% | ||||
| × | なし | ~1% | 6% | ||||
| × | あり | Variable2 | Variable2 | ||||
| × | あり | 50% | 50% | ||||
- 他に罹患した同胞がいない場合。
- 単眼性のretinoblastomaの家系では浸透率が大きく異なる
発端者の他の家族
- 他の家族メンバーのリスクは患者の親の遺伝的状況による。
- 親がRB1遺伝子の生殖細胞系列の変異を持つならば、その家族メンバーはリスクがある。
遺伝カウンセリングに関連した問題
早期の診断と治療を目的としたリスクのある親族への対応は、マネジメントとリスクのある親族への対応を参照
リスク推定
リスクのある成人で症状のない親族のリスク推定のためには、家系内のRB1遺伝子の病的変異が明らかになっていることが必要である。
明らかな新生突然変異による病的変異を持つ家系における注意点
遺伝性Rbの発端者の両親がどちらも病的変異を持たなく、臨床的に罹患していない場合、RB1遺伝子の病的変異は新生突然変異によるものである。しかし、医学的ではない理由によることも考えられる、例えば、父親か母親が違う(生殖補助医療による場合など)、養子縁組を届け出ていない場合などである。
遺伝的発がんリスクとカウンセリング
分子遺伝学的検査を受けるか受けないかに関わらず、リスクのある人における、発がんリスクの評価における、医学的、心理社会的、倫理的問題に関する包括的な記述については、cancer genetics risk assessment and counseling-for health professionals (part of PDQ®, National Cancer Institute)を参照。
家族計画
- 遺伝的なリスクを決定し、出生前検査の実施可能性を議論するのに最適なタイミングは、妊娠前である。
- 罹患しているかリスクのある若い 血縁者には遺伝カウンセリング(子供への遺伝の可能性と生殖に関するオプションに関する議論を含めて)を推奨する[Dommering et al 2012a]。
出生前検査
Rbの家族歴が認められた場合、様々なリスクのある妊娠におけるマネジメント方法が可能である[Canadian Retinoblastoma Society 2009]。
- 罹患した家族のRB1遺伝子の病的変異が判明しているかまたは連鎖解析において有意な情報が得られているならば、リスクのある妊娠に対しては、RB1遺伝子の検査または出生前検査を行っているラボで出生前検査を受けることができる。
- RB1遺伝子変異が胎児に見いだされたら、通常のサイズの眼内腫瘍を発見するために胎児超音波検査を行うこともある。もし発見されれば、早期治療のために早期産で分娩させるという選択肢もありうる[Sahgal et al 2006]。超音波で腫瘍が観察されない場合にも、RB1遺伝子に病的変異をもつ新生児の30%は視力を脅かしうる微細な腫瘍を持つことから、36週での出産が推奨される。
- 家系におけるRB1遺伝子の病的変異がわかっていない場合、出生前超音波検査またはMRIによって罹患した胎児における比較的大きなRbを検出しうるが、こうした検査は、微小なRb腫瘍を検出するほど高感度ではない。
検査の目的が早期診断(そして早期治療)ではなく妊娠中絶である場合には、医療専門家と家族の間に出生前検査の使用についての認識の違いがあるかもしれない。ほとんどのセンターで出生前検査をするかどうかは両親が選択するべきものと考えているが、このような事例では慎重な議論を行うべきである。
着床前診断
RB1遺伝子の病的変異を持つ家系では着床前診断も選択肢になりうる。
関連情報
GeneReviewsスタッフは患者とその家族の利益のため、疾病ごとのあるいはサポート組織として以下に示すものをセレクトした。
GeneReviewsは他の組織が提供する情報に責任を持つものではない
- IRIS Medical
retinoblastoma.com - Childhood Eye Cancer Trust (CHECT)
The Royal London Hospital, Whitechapel Road, London E1 1BB, United Kingdom
Phone: +44 020 7377 5578
Fax: +44 020 7377 0740
Email: info@chect.org.uk
www.chect.org.uk - National Library of Medicine Genetics Home Reference
Retinoblastoma - National Retinoblastoma Parents Group
PO Box 317, Watertown MA 02471
Phone: 800-562-6265
Fax: 617-972-7444
Email: napvi@perkins.pvt.k12.ma.us - NCBI Genes and Disease
Retinoblastoma - American Childhood Cancer Organization (ACCO)
PO Box 498, Kensington MD 20895-0498
Phone: 800-366-2223 (toll-free); 301-962-3520
Fax: 301-962-3521
Email: staff@acco.org
www.acco.org - National Cancer Institute (NCI)
6116 Executive Boulevard, Suite 300, Bethesda MD 20892-8322
Phone: 800-4-CANCER
Children with Cancer: A Guide for Parents - National Federation of the Blind (NFB)
200 East Wells Street (at Jernigan Place), Baltimore MD 21230
Phone: 410-659-9314
Fax: 410-685-5653
Email: pmaurer@nfb.org
www.nfb.org - eyeGENE®- National Ophthalmic Disease Genotyping Network Registry
Phone: 301-435-3032
Email: eyeGENEinfo@nei.nih.gov
eyeGene
分子遺伝学
GeneReviews ではMolecular Genetics と OMIM tablesの情報とは一部異なる可能性がる。最新の情報はOMIM tablesを参照
Table A.
Retinoblastoma: Genes and Databases
| Gene Symbol | Chromosomal Locus | Protein Name | Locus Specific | HGMD |
|---|---|---|---|---|
| RB1 | 13q14?.2 | Retinoblastoma-associated protein | RB1 database rb1-lsdb |
RB1 |
データは下記の基準を参照して編集した。遺伝子表記はHGNC、染色体上の位置,遺伝子座名、相補グループOMIM、タンパク質名はUniProtから。
Table B.
OMIM Entries for Retinoblastoma (View All in OMIM)
| 180200 | RETINOBLASTOMA; RB1 |
| 614041 | RB1 GENE; RB1 |
分子遺伝学的病理
ごくまれに、正常なRB1アレルを持つ細胞から腫瘍化が開始される[Rushlow et al 2013](図1参照)。
遺伝子の構造
27エクソンが転写され、4.7kbのmRNAにスプライスされる。他の機能しうるスプライス構造はない。よく使用される参照配列はNM_000321.2.である。遺伝子とタンパク質の詳細な情報については、TableAのGeneSymbolを参照。
病的ではない変異アレル
2.7kbのORF中に多型が多くみられる部分はないが、イントロンの変異と2つの高度に多型を持つマイクロサテライト(Rb1.20, Tbi2)とミニサテライト(RBD)を有する。
病的変異アレル
網膜芽細胞腫の患者の白血球DNAまたは腫瘍DNAから2500以上の一塩基変異が検出されており、そのうち1700について記録されている(Table A, Locus Specific参照)。RB1の病的変異のほとんどは一塩基変異によって早期の終止コドンを出現させるものが多く、その他にフレームシフト変異や、スプライス変異によってエクソンスキッピングを生じるものがある。病的変異はRB1遺伝子のエクソン1から25とプロモーター領域に散在している。1家系においてエクソン27に病的変異と思われる変異が検出されている[Mitter et al 2009]。病的変異はCGAコドンの一部のメチル化されたCpGジヌクレオチドまたはイントロン12のスプライスドナー部位にも認められた。他の重要な変異は大規模な再構成と欠失である[Albrecht et al 2005, Rushlow et al 2009, Castéra et al 2013]。
通常の遺伝子産物
RB1遺伝子は細胞周期調節(G1期からS期への移行)機能をもつ核タンパク質をコードし、普遍的に発現されている。RBタンパク質はS期への移行の際にCyclin-dependent kinase (cdk) 系因子によってリン酸化される。リン酸化によってポケット部分への結合活性が失われ、細胞タンパク質をリリースする。レビューはDick & Rubin [2013] and Dimaras et al [2015]。
異常な遺伝子産物
RB1遺伝子の病的変異は細胞周期調節機能を失ったタンパク質を発現している。低浸透率の網膜芽細胞腫に関連する変異アレルでは部分的な活性の保持がみられる[Lohmann et al 1994, Bremner et al 1997, Otterson et al 1997]。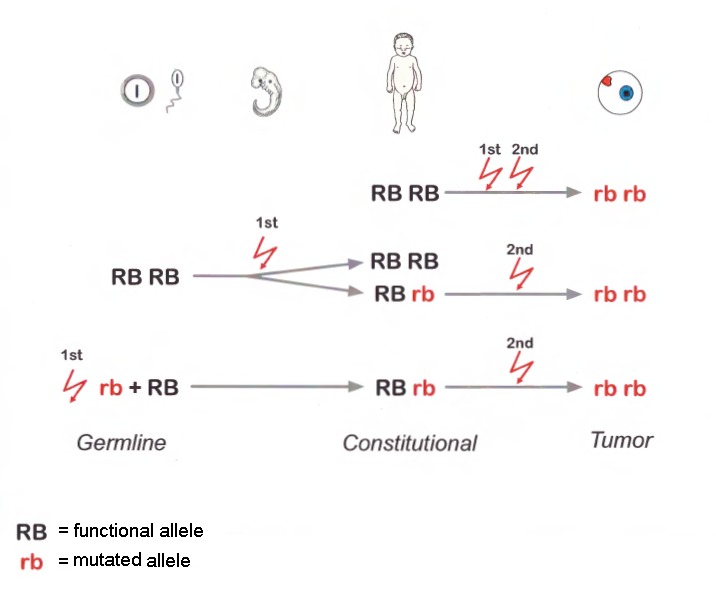
図1
遺伝性と散発性の網膜芽細胞腫(Rb)の分子遺伝学的機構の模式図。RbはRB1遺伝子の両アレルが変異したところから始まる。
散発性のRbでは、二つの変異(最初と2番目)の変異は体細胞で起こる(体細胞変異)
注)体質性細胞(例;末梢血)DNAでは変異は検出されない(2つとも正常アレルRB RB)
遺伝性のRbでは、2番目の変異のみが体細胞性変異である。それぞれの2番目の変異がそれぞれの病巣での腫瘍を誘発する(多巣性Rb腫瘍)。1番目の変異は生殖細胞系列で遺伝する(生殖細胞での新たな変異であっても親から受け継いだ変異であっても)
注)罹患者の体質性細胞では、変異はヘテロである(Rb rb)
時に、罹患児の最初の病的変異は受精後の胚の成長期に起こることもある。
注)罹患児は最初の病的変異を体細胞モザイクで持つ。最初の病的変異が起こった細胞が属する幹細胞系列の細胞から腫瘍が発生する。
更新履歴
-
GeneReview 著者:Dietmar R Lohmann, MD; Brenda L Gallie, MD.
日本語訳者: 福島久代(札幌医科大学大学院修士課程遺伝カウンセリングコース),櫻井晃洋(札幌医科大学 医学部遺伝医学)
GeneReview 最終更新日: 2013.3.28. 日本語訳最終更新日: 2015.11.17 - GeneReview 著者:Dietmar R Lohmann, MD; Brenda L Gallie, MD.
日本語訳者: 福島久代(札幌医科大学大学院修士課程遺伝カウンセリングコース),
櫻井晃洋(札幌医科大学 医学部遺伝医学)
GeneReview 最終更新日: 2015.11.19 日本語訳最終更新日: 2016.9.7 [in present]
![]()