ラッセル・シルバー症候群
(Russell-Silver Syndrome)
[Synonym: Silver-Russell Syndrome]
Gene Reviews著者: Howard M Saal, MD, Director of Clinical Genetics, Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Professor of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, Ohio, gro.cmhcc@mhlaas
日本語訳者: 菅原宏美 田村和朗(近畿大学大学院総合理工学研究科 理学専攻 遺伝カウンセラー養成課程)
Gene Reviews 最終更新日: 2011.6.2.日本語訳最終更新日: 2017.8.10.
要約
疾患の特徴
ラッセル・シルバー症候群(RSS)は、子宮内胎児発育遅延と出生後の成長障害を特徴とする。罹患乳児の出生体重は一般に平均-2SD以下で、出生後の成長については、身長が平均-2SD以下である。罹患者は、典型的には、均整のとれた低身長、正常頭囲、第5指弯曲、額が広く顎が狭い特徴的な三角形の顔、罹患側の成長障害(片側性発育不全)による非対称な四肢長を有する。RSS罹患児の小児期の成長速度は正常である。成人の平均身長は、男性151.2 cm、女性 139.9 cmである。RSSに罹患した小児は、発達遅滞(運動および知的発達)と学習障害のリスクが高いというエビデンスがある。
診断・検査
RSSは遺伝学的に異質性の高い疾患であり、罹患者の多くは、疾患というよりは共通の表現形を呈する。このため診断は、基本的に、共通する臨床的特徴、特に正常頭囲を有する出生前と出生後の発育遅延に基づいて行われる。染色体11p15.5にある父親由来インプリンティング・センター1(IC1)の低メチル化が、RSS罹患者の35-50%に同定される。RSS罹患者の約10%は、7番染色体の母親由来片親性ダイソミー(UPD7)を有する。
臨床的マネジメント
症候の治療:
成長ホルモン治療、運動療法、作業療法、言語聴覚療法、個別の教育計画などが行われる。胃食道逆流症の治療は、体位療法と増粘食で開始し、加えて酸分泌抑制剤(プロトン・ポンプ阻害薬またはラニチジン)の使用が推奨されている。外科治療として、噴門形成術が必要になる場合がある。下肢長が左右で3cm以上違う場合には介入が必要であり、小児期後期に仮骨延長術または骨端軟骨閉鎖術を検討する。重度の小顎症または口蓋裂に対しては多職種の頭蓋顔面専門家チームで治療を行う。停留精巣のある男児は泌尿器科に紹介する。精巣固定術が必要になる場合がある。小陰茎の男児は内分泌科に紹介する。男性ホルモン治療の適応となる場合がある。
サーベイランス:
成長速度のモニタリング、乳児および幼児(必要な場合)に対し低血糖を発見するための血糖値モニタリング、乳児期より健診時の四肢長測定(左右非対称な成長のエビデンスとなる)、言語発達のモニタリング。
遺伝カウンセリング
RSSには複数の病因がある:染色体11p15.5のインプリンティング領域の遺伝子発現を修飾するエピジェニックな変化、母親由来7番染色体の片親性ダイソミー(UPD7)、稀ではあるが常染色体優性または劣性遺伝など。父親由来IC1の低メチル化または母親性UPD7が原因でRSSを発症した発端者の両親はどちらも非罹患と考えられ、同胞の再発リスクは一般集団より大きくはなく、子のリスクもおそらく低い。多くの場合、RSSの発症は家族内で1人だけのため、大部分の妊娠で本疾患のリスクの増加は認められない。このため、通常RSSの出生前診断は実施できない。妊娠中に胎児の超音波検査で子宮内胎児発育遅延が認められた場合には、父親由来H19-IGF2 IC1の低メチル化と母親性UPD7についての出生前検査が可能である。
注意:子宮内胎児発育遅延は、しばしば第3トリメスターまで診断されないことがある。
診断
臨床診断
RSSの臨床診断は、出生後の発育障害を伴う子宮内胎児発育遅延の存在により行う[Silver et al 1953, Russell 1954, Price et al 1999]。RSSには疾患特異的な兆候や特徴はない。
いくつかのRSS診断補助スコアが開発されてきたが、最近では、Netchine et al [2007]とBartholdi et al [2009]が、11p15.5の低メチル化によるRSS患者の表現形に着目して研究を行った(「検査」の項参照)。さらに、多くのRSS患者は、典型的な臨床症状を欠き、より捉えにくい表現形を持つ。Eggermann et al [2009]の研究では、11p15.5のエピ変異を持つRSS患者では、通常より軽度の発育遅延、非対称性、突出した額が認められた。Wakeling et al [2010]も、RSSの臨床症状は、すべての患者で一致したものでないことを示した。
下記の診断基準は、Netchine et al [2007]、Bartholdi et al [2009]、Eggermann et al [2009]およびWakeling et al [2010]の研究から得られた情報を編集したもので、主要項目3項目、または主要項目2項目と副項目2項目に該当する場合には、RSSの診断および裏付け検査を考慮すべきとされる。
- 主要項目
- 子宮内胎児発育遅延/SGA (10パーセンタイル未満)
- 生後の成長で、身長が3パーセンタイル未満
- 正常頭囲(3-97パーセンタイル内)
- 四肢、体幹、顔の非対称性のいずれかまたは複数
- 副項目
- 短い指端距離(上腕と前腕の比率は正常)
- 第5指弯曲
- 三角形の顔
- 前額突出/目立つ額
- 補助項目
- フェオレ斑または皮膚の色素性変化
- 泌尿生殖器異常(停留精巣、尿道下裂)
- 運動、会話、知的発達の遅延のいずれかまたは複数
- 摂食障害
- 低血糖
RSSは遺伝学的に不均一な疾患であり(「検査」の項参照)、一致はしているが変動の大きい形質を示す。RSSの罹患児では、成長ホルモン治療への反応性、追いつき成長、発達にばらつきがみられる。
検査
RSSの原因として、染色体11p15.5に関連するRSSと、7番染色体に関連するRSSの2つが知られている。
染色体11p15.5関連RSSは、染色体11p15.5のインプリンティング領域の変化により起こる[Abu-Amero et al 2010]。染色体11p15.5領域には、胎児および胎盤の成長に重要な役割を持つインプリンティング遺伝子のクラスターがある。遺伝子のインプリンティングとは、両親から受け継いだ1組の遺伝子のDNAがそれぞれ異なる修飾を受け、片方の遺伝子のみが発現する現象である。両親のどちらの遺伝子が発現するかは、個々のインプリンティング遺伝子によって決まっている。
インプリンティング遺伝子は、しばしばインプリンティング・センター(IC)を含むクラスターとしてみられる。11p15.5のインプリンティング・クラスターのひとつには、インプリンティング・センター1(IC1)があり、胎児の発達に重要な成長因子をコードするIGF2遺伝子と、非翻訳転写産物をコードする H19遺伝子の発現を相互的に調整しており、IC1は、父親由来と母親由来で異なるメチル化を受けている(図1A)。RSSでは、IC1の低メチル化により、H19の発現とIGF2の発現抑制が両方のアレルで起こることにより、成長障害が起こる(図1B)。
「分子遺伝学的病因」の項参照。
図1 染色体11p15.5関連RSS
インプリンティング・クラスターの図解
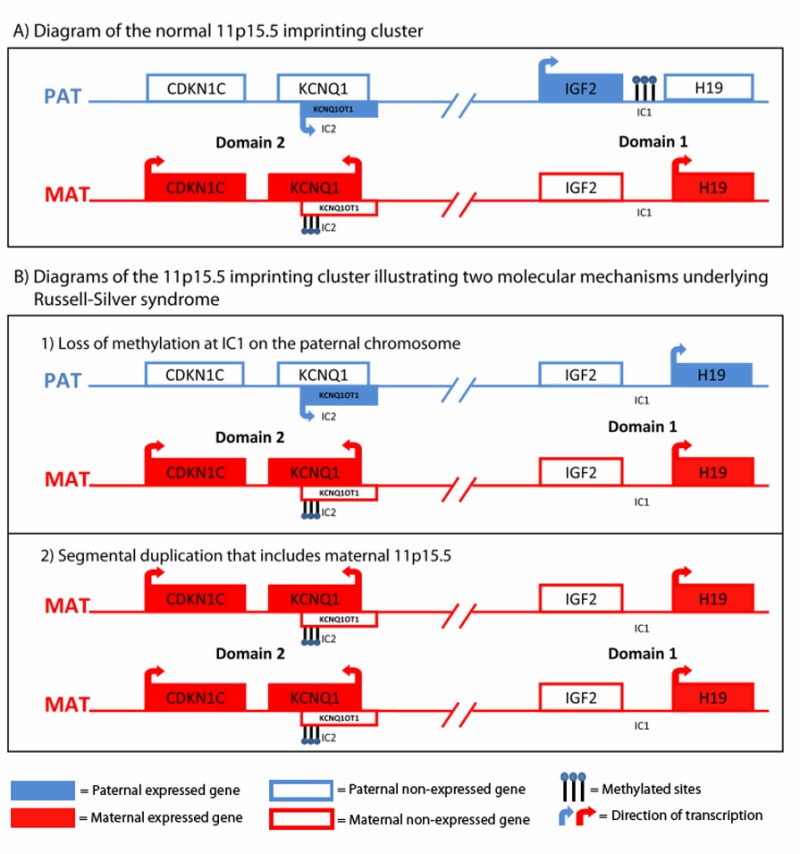
図1A
染色体11p15.5のインプリンティング・クラスターは機能的に2つのドメインに分けられる。
ドメイン1
ドメイン1の調節不全は、11p11.5関連RSSに関係している。ドメイン1には、2つのインプリンティング遺伝子がある。
1) IGF2:インスリン様成長因子をコード
2) H19:非翻訳RNAをコード
H19に関連するインプリンティング・センター(IC1)は、通常、父親由来の染色体上ではメチル化され、母親由来染色体上ではメチル化されていない。すなわち、父親由来アレルではIGF2が発現し、母親由来アレルではH19が発現するよう調節されている。
ドメイン2
ドメイン1とドメイン2両方の機能不全は、Beckwith-Wiedemann症候群に関係している。ドメイン2にはCDKN1C、KCNQ1、KCNQ1OT1といったインプリンティング遺伝子が含まれる。インプリンティング・センター2(IC2)はKCNQ1OT1のプロモーターを含む。KCNQ1OT1の非翻訳転写産物は、同アレル上のドメイン2のインプリンティング遺伝子の発現を調整する。IC2は、通常、母親由来染色体上でメチル化され、父親由来染色体上ではメチル化されていない。すなわち、母親由来アレルではCDKN1CとKCNQ1が発現し、父親由来アレルでKCNQ1OT1が発現するよう調節されている。
図1B
インプリンティングの変化によりRSSが発症する2つの例
1)父親由来IC1のメチル化の消失がRSS患者の30-50%にみられる。このエピジェネティックな変化により、H19の発現と、成長因子をコードするIGF2の発現消失が両方のアレルで起こる。
2)母親由来11p15.5を含む領域の部分的な重複が、少数のRSS患者でみられる。重複は、細胞学的検査/FISHで検出できる大きさのものもあれば、分子学的な重複/欠失解析を必要とするもっと小さいものもある。母親由来11p15.5のダイソミーでRSSが起こるメカニズムは不明であるが、上流のCDKN1Cインプリンティング・クラスターにある遺伝子の量的効果が関与する可能性がある[Fisher et al 2002]。
Weksberg et al [2010]; Macmillan Publishers Ltd.の許諾を受けて掲載
メチル化解析
- 父親由来染色体のIC1の低メチル化が、RSS患者の30-50%で検出される(図1B)。IC1はIGF2とH19のメチル化を制御するため、遺伝子別の解析では、多くの場合、両方の遺伝子で低メチル化を示す。
- 父親由来IC1のある11p15.5の低メチル化は接合子形成後に起こるため、多くのRSS患者では、メチル化状態の異なる複数のパターンが体細胞に分布する(表1 「検査結果の解釈」参照)。
- 少数のRSS患者ではH19またはIGF2だけが選択的に低メチル化されている[Bartholdi et al 2009]。
- 少数のRSS 患者ではIGF2R(染色体6q25-q27上のIGF2受容体をコードする遺伝子)の高メチル化がみられ、H19のメチル化は正常である[Turner et al 2010]。これらの例では、循環血液中のIGF2を除去して成長効果を制限しているIGF2受容体が減少することによってRSSが発症したと考えられる[Braulke 1999]。
欠失/重複解析
- 少数のRSS患者では、母親由来染色体11p15.5領域の重複がみられる。転座や逆位に伴ってみられるサイズの大きい重複は、細胞遺伝学的解析によって検出できる[Fisher et al 2002, Eggermann et al 2005]が、より解像度の高い欠失/重複検査法の方が、感度が高い(表1)。母親由来11p15.5のダイソミーでRSSが起こるメカニズムは不明であるが、上流のCDKN1Cインプリンティング・クラスターにある遺伝子の量的効果が関与する可能性がある[Fisher et al 2002]。
- インプリンティング・センター2(IC2)の重複が母親から受け継がれた例が1人のRSS患者で確認されている[Schönherr et al 2007]。この発見の影響およびRSS患者数への寄与は不明であり、さらなる研究が待たれる。
7番染色体関連RSS
- 母親に由来する7番染色体の片親性ダイソミー(UPD7)がRSS 患者の7-10%にみられる[Moore et al 1999, Hannula et al 2001, Kim et al 2005]。しかしながら、UPD7インプリンティングに関連する特別の遺伝子座は明らかになっていない(「分子遺伝学的病因」の項参照)。
・ 母親性アイソダイソミーと母親性ヘテロダイソミーが報告されている[Bernard et al 1999, Price et al 1999]。- UPD7のモザイク例がみつかっている[Reboul et al 2006]。
- 部分的なUPD7がみつかっている:Hannula et al [2001]は、7q31-qter領域の母親性UPDの1例を報告し、Eggermann [2008]は7番染色体の長腕の大部分(7q11.2-qter)のUPDを2例報告している。
- RSS患者でみられる稀な7番染色体構造変化としては、以下のものがある。
- 母親由来7番染色体のヘテロダイソミーを持つ2人の小児で、7番染色体のトリソミー・モザイクが報告されている[Flori et al 2005, Font-Montgomery et al 2005]。うち1人は出生前に確認された[Font-Montgomery et al 2005]。
- 7番染色体長腕の中間部欠失[del(7)(q21.1q21.3)]が1人の小児でみつかっている[Courtens et al 2005]。
- 7p11.2-p12の微細な重複が、FISH法で同定された(通常の核型検査では検出できないため、FISH法または別の欠失/重複解析が必要とされる。表1参照)[Joyce et al 1999, Monk et al 2000]。
表1 RSSの分子遺伝学的検査
| RSSのタイプ | 遺伝学的機構 | 検査方法 | 検出される変異や変化1 | RSSの割合2 |
|---|---|---|---|---|
| 染色体11p15.5関連RSS | 父親由来11p15.1のIC1メチル化消失 | 11p15.5メチル化解析 | 父親由来IC1の低メチル化3,4 | ~35-50% |
| 母親由来11p15.5の重複 | 重複/欠失解析5 | 11p15.5の重複 | 不明 | |
| 7番染色体関連RSS | UPD(母親由来) | UPD解析(種々の方法による)6 | 7番染色体の母親由来ダイソミー7 | ~7-10% |
| 欠失/重複 | 欠失/重複解析、細胞遺伝学的解析 | 7番染色体の構造変化 | 稀 |
- 検出される変異/変化については「分子遺伝学」の項を参照されたい。
- 遺伝学的機構と染色体位置に基づいて使用された検査法での変化の検出力。
- 11p15.5の低メチル化は、受精後に起こるため、体細胞モザイクによる偽陰性が起こりうる。別の組織(口腔粘膜細胞や線維芽細胞など)の検査を行うべきである。
- 特定の遺伝子座のメチル化が胎芽期のどのタイミングで起こるかは不確実であるため、出生前診断目的での11p15.5のメチル化解析は推奨されない。
- 翻訳領域およびイントロン近傍の配列解析では検出できない、エクソンや遺伝子全体の欠失の検査法。定量的PCR法、long-range PCR法、MLPA法、この遺伝子/染色体を含む領域の染色体マイクロアレイ検査などさまざまな方法がある。
- UPDの検出にはさまざまな方法がある。例えば、SNPまたはマーカー解析、メチル化特異的MLPA法など。検査には、両親の血液検体が必要である。
- 母親性UPDおよびその他の7番染色体構造変化では、体細胞モザイクの存在が知られている。別の組織を使った検査が適切な場合がある。
検査の進め方
RSSの診断を確定するための発端者の検査は、以下の順番で行うことが推奨されている。
- 臨床背景からRSSが疑われる場合、11p15.5のIC1メチル化解析とUPD7の検査を同時にオーダーするのがもっとも効果的である。メチル化解析により、11p15.5の重複や欠失も検出できる。11p15.5領域に対しては、MS-MLPAがもっとも堅牢な検査方法であり、メチル化の変化、UPD11、欠失/重複の3種を検出することができる。リンパ球を使った検査が陰性であれば、体細胞モザイクの可能性を考え、ほかの組織(口腔粘膜細胞など)での再検査を考慮する。
- 臨床的にRSSの可能性が低い場合には、アレイ・ハイブリダイゼーション法による欠失/重複解析を最初に行うべきである。
分子遺伝学的検査のアルゴリズムが、Eggermann et al [2010]により論文発表されている。
臨床的特徴
臨床像
重要な臨床的特徴[Price et al 1999]:
- 子宮内胎児発育遅延(IUGR):出生体重が平均の-2SD以下
- 出生後の成長障害:身長が平均の-2SD以下
- 頭囲は正常で、しばしば「偽水頭症」にみえる
- 第5指弯曲
- 四肢長が非対称
診断の補助となり得るその他の特徴
- 低身長で上半身と下半身の比率が正常、骨格変化なし、骨年齢の遅れ(高頻度)
- 広く目立つ額、小さい三角形の顔、小さく狭い顎、下向きの口角など顔の特徴
- 低血糖
- 短指症、屈指症
- カフェオレ斑
- 指端距離が身長より短い
成長
RSS罹患児の初期の問題は、成長と栄養補給に関するものである。RSS罹患児は、子宮内胎児発育遅延と出生後の成長障害を示す。成長指標として、欧州のRSS児用成長曲線が発表されている[Wollmann et al 1995]。北米のRSS児の成長曲線は、MAGIC Foundationから入手できる。
成長速度は正常である。成長ホルモン治療を受けないRSS患者で、成人男性の平均身長は151.2 cm(-7.8 SD)、成人女性は139.9 cm(-9 SD)である[Wollmann et al 1995]。
多くのRSS患者で、身長に対して短い指端距離(上腕と前腕の比率は正常)が認められるものの、均整のとれた成長が期待できる[Silver et al 1953, Saal et al 1985]。
RSS患児への成長ホルモン治療の効果については、「治療」の項参照。
注意:成長ホルモン治療を行っても正常身長を達成できないRSS患は多い。
RSSと診断され、小児期の遅くに追いつき成長を示す患者の多くは[Saal et al 1985]、古典的なRSSとは違っていた可能性がある。
成長ホルモン分泌不全
24人のRSS患児を対象とした研究で、10人に低血糖がみられた。成長ホルモン分泌不全(グルカゴン刺激試験による)が何人かでみつかり、1人は低血糖の原因と考えられた[Azcona & Stanhope 2005]。
骨格初見は、RSSの場合、通常、四肢長の非対称性に限定され、少なくとも何人かの患者でみられている。これは罹患側の成長の遅れによる片側性発育不全と考えられる。
診断基準として使われているように、第5指弯曲は、RSS患者でもっとも頻度の高い骨格初見である。
25人のRSS患者を対象とした整形外科学的兆候の系統的な研究で、19人に中手骨と指骨の変化、9人に側弯、5人につま先の合指症、3人に骨盤の発生学的異形成が認められた[Abraham et al 2004]。
神経発達
成長の問題を除けば、神経発達は、両親がもっとも関心を持つ事項である。初期の報告ではRSS患者の「知能は正常」とされ安心材料になっていたが、この病気の小児は、発達遅滞(運動発達と知的発達の両方)および学習障害のリスクが高いとするエビデンスが増加しつつある。
- 6歳から12歳のRSS患児20人を対象とした研究で、平均IQは86であった。また、36%は特別な教育、48%は言語聴覚療法を必要としていた[Lai et al 1994]。RSSの病因が明らかになっている児はいなかった。
- 36人のRSS患児を対象とした別の研究で、平均IQは患児群で95.7、対照同胞群で104.20であった。特記すべき点として、母親性UPD7の患児2人のIQはそれぞれ81と84であった[Noeker & Wollmann 2004]。
- 11p15の低メチル化と母親性UPD7の両方を含む大規模コホートの再評価で、軽度の発達遅滞は、11p15低メチル化群に比べてUPD7群で多かった(65% vs 20%)。会話の遅れは、両グループで共通にみられた[Wakeling et al 2010]。
低血糖
RSSの患児は皮下脂肪が少なく、極度に痩せており、しばしば食欲不振を有するため、手術時など食事間隔の延長によって低血糖を起こしやすい[Tomiyama et al 1999]。RSS患児を対象とした研究で、低血糖に関連する因子には、カロリー摂取量の減少(食欲不振や摂食不良に続発することが多い)、体重の減少、一部の患児では成長ホルモン分泌不全があった[Azcona & Stanhope 2005]。多くの患児が低血糖の臨床症状(特に多量の発汗)を有した一方で、一部に無症状の者もあった。
多汗症は低血糖がなくとも起こるが、小児期早期の多汗症は、低血糖と関連している可能性がある[Stanhope et al 1998]。
消化器症状がよくみられる[Anderson et al 2002]。胃食道逆流症、食道炎、食物嫌悪、成長障害などの問題がみられる。いくつかは医原性の可能性がある(成長の遅れに対する治療に関連)。食物嫌悪や誤嚥がみられる児では、逆流性食道炎が疑われる。
重度の頭蓋顔面初見は一般的でない。一部のRSS患者は、ピエール・ロバン・シークエンスおよび口蓋裂を有する。Wakeling et al [2010]は、11p15.5低メチル化の患者の7%に口蓋裂または口蓋垂裂が認められたが、母親性UPD7の患者にはみられなかったとしている。
歯と口腔の初見は稀である。矮小歯、高口蓋、小顎症と小さな口に伴う二次的な歯の密生が報告されている[Cullen & Wesley 1987, Kulkarni et al 1995, Orbak et al 2005, Wakeling et al 2010]。
歯の密生があると口腔衛生が悪くなり、齲歯のリスクが高くなる。
泌尿生殖系の問題は報告されているが、一般的でない。もっともよくみられるのは尿道下裂と停留精巣である。腎疾患としては、水腎症、尿細管性アシドーシス、後部尿道弁、馬蹄腎が報告されている[Arai et al 1988, Ortiz et al 1991]。
新生物
ウイルムス腫瘍、肝細胞がん、頭蓋咽頭腫などの新生物の報告が時折あるが、RSS患者で新生物の発症頻度が増加することはない[Draznin et al 1980, Chitayat et al 1988, Bruckheimer & Abrahamov 1993]。
遺伝子型と臨床型の関連
メチル化特異的制限酵素HpaIIまたはNotIを使ってH19のメチル化の程度を測定することにより、Bruce et al [2009]は、H19メチル化高度減少、H19メチル化減少、H19メチル化正常、母親性UPD7(H19メチル化正常)に分類するスケールを開発した。H19メチル化高度減少(メチル化の程度が -6SD以下、または9%未満)の患児は、H19メチル化減少および母親性UPD7の患児に比べ、より重度の骨格症状(橈骨上腕骨脱臼、合指症、四肢の非対称性大、側弯など)を有する傾向がみられた。
Wakeling et al [2010]の研究では、IC1低メチル化によるRSS患児と母親性UPD7によるRSSの臨床的特徴を比較した。両者には、表現形の重なりがかなり認められた。第5指弯曲と認知機能障害はIC1低メチル化患児でより頻度が高く、一方学習障害と会話の問題は、母親性UPD7の患児でより高頻度にみられた。
ウイルムス腫瘍や肝芽腫、Beckwith-Wiedemann症候群患者でみられるその他の腹部腫瘍に関連する染色体11p15.5のインプリンティング領域の変異を、少なくとも一部のRSS患者は持つことを考えると、RSS患者で悪性腫瘍のリスクが低いことは重要である。腫瘍リスクは、過成長に伴う変異があると上昇し、逆に成長遅延に関わる変異では減少するようにみえる。
頻度
RSSの有病率は10万人に1人と推計される[Christoforidis et al 2005]。
遺伝学的関連(アレル)のある疾患
Beckwith-Wiedemann症候群は、染色体11p15.5(BWS責任領域としても知られる)のインプリンティング領域の遺伝子転写制御の変化により起こる。
IC2の低メチル化、IC1の高メチル化[Martin et al 2005]、11p15の父親性UPD [Shuman et al 2002] など、11p15の異なる分子機構が、片側性過形成症の孤発例で報告されている。
父親由来IC1メチル化欠失の体細胞モザイクが、孤発性片側性低形成症で報告されている[Zeschnigk et al 2008, Eggermann 2009]。
ウイルムス腫瘍の孤発例には、IC1の高メチル化、11p15.5の父親性UPD、微細欠失や微細挿入を含むゲノム変化など、生殖細胞系列の染色体11p15.5変化が関連することがある[Scott et al 2008]。
鑑別診断
子宮内胎児発育遅延と低身長
ラッセル・シルバー症候群(RSS)の鑑別診断として、子宮内胎児発育遅延と低身長の原因となるあらゆる疾患が対象となる。
染色体疾患および欠失/重複解析
染色体の不均衡で起こる多くの疾患がRSSと誤って診断される可能性があるため、RSSと同様の所見を持つ小児に対しては、通常の細胞遺伝学検査よりも検出力の高い染色体検査(アレイCGHとSNPマイクロアレイを含む高解像度の染色体マイクロアレイが好ましい)を実施すべきである。
RSSの鑑別診断として考慮すべき染色体疾患には次のものがある。
- Y染色体長腕欠失[Leppig et al 1991]
- 二倍体と三倍体の倍数性混在(四肢の非対称性がみられる)[Graham et al 1981]
- ターナー症候群モザイク[Li et al 2004]
- 12p14欠失が、RSSのいくつかの特徴を持つ小頭症と知的障害の患者で検出された[Spengler et al 2010]
- 15q26.3(IGF1Rを含む)の欠失および22q11.2の遠位欠失(子宮内胎児発育遅延との関連が知られている)[Spengler et al 2010]。
- 17q25の再配列[Ramírez-Dueñas et al 1992, Midro et al 1993]。
DNA修復障害による疾患
ファンコニ貧血、ナイミーヘン症候群、ブルーム症候群ではしばしば子宮内胎児発育遅延と低身長がみられる。これらの疾患では、通常、小頭症、皮膚の日光過敏、四肢の奇形などの臨床的特徴が明確である。
その他
- RSSと混同されてきた疾患の一つに、X連鎖性の皮膚色素過剰を伴う低身長症がある。Partington [1986]が最初の症例を報告し、これをX連鎖性RSSと呼んだ。この疾患は、家族歴がない場合は古典的なRSSと区別するのが難しいと思われる。
- 3-M症候群は、出生前と出生後の成長遅延、特徴的な顔貌(相対的大頭、前頭隆起、尖った目立つ顎、肉づきがよく上を向いた鼻、豊かな唇と眉、顔面中央部の低形成)、放射線学的所見を特徴とする。知能は正常。最終身長は平均より5-6SD低い。特徴的な放射線所見として、細長い骨、痩せた四肢、経年的に縮んでいく高い椎体、潜在性二分脊椎、小さい骨盤、小さい腸骨翼、骨年齢の遅れがみられる。CUL7の変異が原因で、常染色体劣性遺伝形式をとる。
- 胎児アルコール症候群(FAS)の児は、一般に子宮内胎児発育遅延、小頭症、成長障害、しばしば三角形の顔を示す。FASの小児の多くは、子宮内エタノール曝露が確認でき、顔の所見(短い眼瞼列、平坦な人中、厚い上唇)が特徴的である。
- IMAGe症候群は、子宮内胎児発育遅延、骨幹端異形成、先天性副腎低形成、停留精巣および小陰茎などの生殖器所見を特徴とする。頭囲は正常である[Vilain et al 1999, Pedreira et al 2004]。IMAGe症候群は、CDKN1C遺伝子の病原性バリアントが母親から伝わることで発症する。
- RSSに類似した骨格異形成症を除外するために、骨格の検索を行うべきである。
注意:RSS患児でも骨年齢が遅れることがある。しかしながら、骨年齢の遅れは、さまざまな原因による子宮内胎児発育遅延の児に高頻度にみられる、非特異的な所見である。
小頭症
RSS 患者の頭囲は正常である。頭囲が平均より3SDを超えて小さい場合には、成長障害を起こす別の原因を探すべきである。
臨床的マネジメント
初回診断後の評価
ラッセル・シルバー症候群(RSS)と診断された患者の疾患の程度を明らかにするため、以下の評価を行うことが勧められる。
- 成長曲線の作成と評価。欧州の小児はWollmann et al [1995]、北米の小児はMAGIC Foundationを参照されたい。
- 診察により、四肢長の非対称性、口腔および頭蓋顔面の特徴的所見がないか評価する。
- 大部分のRSS患児に対して、標準法による成長ホルモン分泌不全の評価を行う。
- 発汗量の多い患児に対して、低血糖の評価を行う。
- 胃食道逆流症(GERD)が疑われる患児に対して、食道造影検査、食道内pH測定、内視鏡などによる食道の評価を行う。
- 神経認知発達、言語、筋緊張のスクリーニング。
症状の治療
成長
身体的な相違点や低身長を伴う疾患の小児は、ボディイメージに敏感であることが多い。これらの要素は、自己イメージ、仲間との関係性、社会化に大きな役割を果たす場合がある。このため、RSSの小児に対する心理カウンセリングが有用であることが多い。
さまざまな原因による子宮内胎児発育遅延の児へのヒト成長ホルモン治療は、成長および最終身長を有意に改善してきた[Albanese & Stanhope 1997, Azcona et al 1998, Czernichow & Fjellestad-Paulsen 1998, Saenger 2002]。とりわけRSSの患児では、成長ホルモン分泌不全がなくても、成長ホルモン補充のベネフィットがあり[Albanese & Stanhope 1997]、有意な成長速度の改善と最終身長の改善が認められ[Azcona et al 1998]、成長ホルモン治療を中止した後も成長速度の改善が継続した[Azcona & Stanhope 1999]。
成長ホルモン補充療法は、成長に関連する疾患の管理経験が豊富な医療機関で行うのが最良である。
成長ホルモン治療を受けたRSS患児では、身長の有意な増加が認められたが、体幹および四肢の非対称性には変化がなかったとする研究がある[Rizzo et al 2001]。
UPD7が原因のRSS患児は、11p15.5低メチル化が原因の患児に比べて、成長ホルモン治療による身長増加が大きかった。これはおそらく11p15.5低メチル化が原因の場合はインスリン様成長因子1(IGF1)のレベルが高いためである。UPD7が原因のRSS患児の成長ホルモン治療に対する反応性は、ほかの原因によるSGAの小児と同じであった[Binder et al 2008]。
IGF1とIGF1結合タンパク-3(IGFBP-3)を検討した最近の研究では、これらのタンパクレベルの変化と、成長ホルモン治療後の成長速度の変化には関連がなかった。しかしながらこの研究では、RSSの診断が臨床所見のみに基づいて行われ、11p15低メチル化や母親性UPD7の検査データは報告されていない[Beserra et al 2010]。
成長ホルモン治療を受けた26人のRSS患児を対象にした、観察期間中央値9.8年の長期アウトカム研究では、研究開始時に-2.7SD(中央値)であった身長が、治療により-1.3SD(中央値)となり有意な効果が報告された[Toumba et al 2010]。
RSSは遺伝学的に不均質な疾患であり、研究に病因のデータが含まれていないと、成長ホルモン治療を受けたRSS患児に関する多くの研究結果を解釈することは難しい。また、RSS患児の成長ホルモン治療に関しては、長期効果の観察が重要で、特に成人の最終身長への影響、および四肢長非対称性のある患者の整形外科的な変化の有無を長期的に評価する必要がある。
成長ホルモン分泌不全
成長ホルモン分泌不全がある場合には、ヒト成長ホルモンによる治療が必要である。
骨格所見
下肢長の左右差が3cmより大きいと、代償性に側弯を生じることがあるため、介入が必要である。補高靴の使用から治療を開始する。年長の小児に対しては、仮骨延長術または骨端軟骨閉鎖術を検討する。
神経発達
- 筋緊張低下がみられる乳児は、早期介入プログラムと理学療法に紹介する。
- 発達遅滞が認められた小児は、早期介入および言語聴覚療法に紹介する。
- 学童期の小児に対しては、学校との協力の上で、適切な神経心理学的検査による学習障害の評価を行い、個別の教育計画を作成する。
低血糖は、栄養補充、食事回数の増加、複合糖質の使用による標準的な方法で治療を行う。
消化器症状(積極な管理を要するもの)
- 胃食道逆流症の治療は、体位療法と増粘食で開始し、加えて酸分泌抑制剤(できればオメプラゾール、パントプラゾールなどのプロトンポンプ阻害薬)の使用が推奨されている。より重症の場合または保存的治療の効果が不十分な場合には、外科治療として、噴門形成術が必要になることがある。
- 食物嫌悪は、言語聴覚士や作業療法士によって療育の中で発見される可能性がある。
頭蓋顔面所見
重度の小顎症または口蓋裂に対しては、多職種の頭蓋顔面専門家チームで治療を行う。下顎矯正手術が必要になることは稀である。
歯科衛生および歯の密生については、小児歯科医と矯正歯科医による通常の方法で管理できる。
泌尿生殖器所見
- 停留精巣のある男児は泌尿器科に紹介する。精巣固定術が必要になる場合がある。
- 小陰茎の男児は内分泌科に紹介する。男性ホルモン治療の適応となる場合がある。
新生物
RSS患者の悪性腫瘍のリスクは低い。体幹の非対称性がある場合でも、Beckwith-Wiedemann症候群でみられるような片側性の過形成ではないため、RSS患児は、定期的な腹部および腎臓の超音波検査の適応とならない。
サーベイランス
以下を実施する。
- 成長速度に着目した成長のモニタリング。
- 多汗または食欲のない乳児および年長の小児に対し、低血糖を発見するための血糖値モニタリング。
- 乳児期からの健診時に、診察および四肢長差の測定。
- 言語発達の綿密なモニタリング。
血縁者のリスクの評価
遺伝カウンセリングの目的については、「遺伝カウンセリング」の項で、リスクのある血縁者の検査に関連した問題を参照されたい。
研究中の治療
ClinicalTrials.govでは、幅広い疾患や状況の臨床研究の情報が得られる。
注:この疾患の臨床試験は登録されていないかも知れない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ラッセル・シルバー症候群(RSS)は複数の病因で起こる:父親由来H19-IGF2 インプリンティング・センター1(IC1)のメチル化の消失、母親由来7番染色体の片親性ダイソミー(UPD7)など。常染色体優性または劣性遺伝形式をとるものは稀である[Ounap et al 2004]。
血縁者のリスク − 染色体11p15.5関連RSS:父親由来H19-IGF2 IC1のメチル化消失が原因の場合
発端者の両親
- 発端者が染色体11p15.5のインプリンティング欠失によるRSSの場合、通常、両親は非罹患である。
- Bartholdi et al [2009]は、H19-IGF2 IC1のメチル化欠失によるRSSの親子例(父親と娘)を報告している。
- 同胞にエピジェネティック変異が確認された2家系で、非罹患父親の性腺モザイクが推定されている[Bartholdi et al 2009]。
発端者の同胞
- 発端者が染色体11p15.5のインプリンティング欠失によるRSSの場合、通常、同胞のリスクは一般集団のリスクより高くはない。
- 染色体11p15.5のインプリンティング欠失が父親から子どもに伝わった例、非罹患父親に性腺モザイクが推定される例が報告されている。このため、家系によっては同胞のリスクが増加することがある。
発端者の子
- 発端者の子のリスクは低いと考えられる。
- 染色体11p15.5のインプリンティング欠失が父親から娘に伝わったとする1編の報告[Bartholdi et al 2009]を除くと、染色体11p15.5のインプリンティング欠失を持つ発端者の再発リスク決定に資するデータはない。
その他の血縁者
発端者が染色体11p15.5のインプリンティング欠失によるRSSの場合、通常、その他の血縁者のリスクは一般集団のリスクより高くはないと考えられる。
血縁者のリスク − 7番染色体関連RSS:母親由来の片親性ダイソミー7
発端者の両親
発端者が母親性UPD7によるRSSの場合、両親は非罹患と言える。
発端者の同胞
発端者が母親性UPD7によるRSSの場合、同胞のリスクは一般集団のリスクより高くはない。
発端者の子
発端者の子のリスクは低いと考えられる。母親性UPD7を持つ発端者の再発リスク決定に資するデータはない。
その他の血縁者
発端者が母親性UPD7によるRSSの場合、通常、その他の血縁者のリスクは一般集団のリスクより高くはない。
遺伝カウンセリングに関連した問題
経験的リスク
11p15の低メチル化または母親性UPD7が同定され、両親と同胞の身長が正常な患者の45-60%で、同胞の再発リスクは増加せず一般集団と同じである。
DNAバンキング
DNAバンキングは、将来使用する可能性に備えてDNA(一般に白血球細胞から抽出したもの)を保存しておくことである。検査方法、および我々の遺伝子、アレルバリアント、疾患に関する理解は将来進歩する可能性があるため、罹患者のDNAバンキングについて考慮すべきである。
出生前診断
多くの場合、RSSの発症は家族内で1人だけのため、大部分の妊娠では本疾患のリスク増加は認められない。このため、通常、RSSの出生前診断は実施できない。
妊娠中に胎児の超音波検査で子宮内胎児発育遅延が認められた場合には、両親および羊水中の胎児細胞を使ったPCR法により、父親由来H19-IGF2 IC1の低メチル化と母親性UPD7についての出生前検査が可能である。
注意:子宮内胎児発育遅延は、しばしば第3トリメスターまで診断されないことがある。
分子遺伝学
分子遺伝学およびOMIMの表の情報は、GeneReviewsの他の部分の記載内容と異なる可能性がある。表には、より最近の情報が含まれていることがある。
表A
ラッセル・シルバー症候群:遺伝子およびデータベース
| 遺伝子 | 染色体座位 | タンパク質 | 遺伝子特異的データベース | HGMD |
|---|---|---|---|---|
| H19 | 11p15?.5 | Unknown | H19 @ LOVD | H19 |
| IGF2 | 11p15?.5 | Insulin-like growth factor II | LOVD - Growth Consortium (IGF2) | IGF2 |
| Unknown | 7番染色体 | Unknown |
データは以下の標準的参照資料をもとに作成した。遺伝子はHGNC、染色体座位、座位の名称、遺伝子変異に密接に関連した領域、相補群はOMIM、タンパク質はUniProtを参照した。遺伝子特異的データベースとHGMDの説明は、こちらを参照されたい。
表B
ラッセル・シルバー症候群のOMIM 登録番号(内容はOMIMを参照されたい)
| 103280 | H19, IMPRINTED MATERNALLY EXPRESSED NONCODING TRANSCRIPT; H19 |
| 147470 | INSULIN-LIKE GROWTH FACTOR II; IGF2 |
| 180860 | ILVER-RUSSELL SYNDROME; SRS |
分子遺伝学的病因
染色体11p15.5関連RSS
染色体11p15.5にあるインプリンティング遺伝子は、胎児の成長に重要であることが知られている[DeChiara et al 1990, Fitzpatrick et al 2002, Eggermann 2009]。RSSはインプリンティング・ドメイン1のエピジェネティックな変化によって発症する[Gicquel et al 2005, 図1]。これに対し、過成長症であるBeckwith-Wiedemann症候群は、インプリンティング・ドメイン1とドメイン2の両方がエピジェネティックな変化を起こすことで発症する(Beckwith-Wiedemann症候群のFigure 1参照)。11p15.5のメチル化領域の違いが遺伝子転写に影響するメカニズムは明確になっていない。IC1とzinc-fingerタンパクであるCTCF(CCCTC-binding factor:クロマチン構造をコントロールする転写制御因子)との結合により、クロマチン・ドメインの活性化と不活化が起こる1つのモデルが提唱されている[Li et al 2008, Demars et al 2010]。RSSでは、ドメイン1の低メチル化によりクロマチン構造が変化し、同じアレルの転写促進配列と制御タンパク質からのシグナルのどちらかまたは両方がブロックされるためにIGF2の転写がOFFになり、H19の発現が両アレルで起こる。RSSの原因としてのH19-IGF2とIC1の研究には、以下のものがある:Obermann et al [2004]、Gicquel et al [2005]、Schönherr et al [2007]、Turner et al [2010]。
- H19は、母親由来遺伝子が発現するインプリンティング遺伝子で、転写産物は2322ヌクレオチドのRNA(非翻訳)である。H19のインプリンティングはIC1ドメイン(図1)により制御される。
- IGF2は父親由来遺伝子が発現するインプリンティング遺伝子で、インスリン・ファミリーに属する成長因子ポリペプチドをコードする。この遺伝子産物は、発達と発育に関与する。もっとも多い転写産物はNM_000612.4で、180個のアミノ酸からなるインスリン様成長因子IIのアイソフォーム1(NP_000603.1)をコードしている。
7番染色体関連RSS
UPD7インプリンティングに関連する特別の遺伝子座は明らかになっていない。しかし、7番染色体長腕(7q)の母親性UPDの症例が複数報告されていることから、関連する遺伝子は7q上にあると考えられる[Eggermann 2008]。Hannula et al [2001]は、7q31-qter領域の母親性UPDの1例を報告している。
更新履歴
-
Gene Reviews著者: Howard M Saal, MD, Director of Clinical Genetics, Division of Human Genetics, Cincinnati Children's Hospital Medical Center, Professor of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, Ohio, gro.cmhcc@mhlaas
日本語訳者: 菅原宏美 田村和朗(近畿大学大学院総合理工学研究科 理学専攻 遺伝カウンセラー養成課程)
Gene Reviews 最終更新日: 2011. 6. 2.日本語訳最終更新日: 2017.8.10.(in present)
![]()