ルビンスタイン・テイビ症候群
(Rubinstein-Taybi Syndrome)
[Synonym: Broad Thumbs-Hallux Syndrome]
Gene Reviews著者: Cathy A Stevens、 MD
日本語訳者:小崎 里華 (国立成育医療研究センター 遺伝診療科)
GeneReviews最終更新日: 2023.11.9. 日本語訳最終更新日:2024.10.14..
要約
疾患の特徴
ルビンスタイン・テイビ症候群 (以下RSTS)は特徴ある顔貌、幅広く、しばしば偏位した母指趾、低身長、中等度~重度の精神遅滞を特徴とする症候群である。顔貌の特徴として、眼瞼裂斜下、鼻翼より下方に伸びた鼻柱(鼻中隔下端)、高口蓋、しかめっつらの笑顔、切歯結節がある。胎児期の成長は正常であるが、生後数ヶ月で、身長、体重および頭囲のパーセンタイルは急速に低下する。成人期には、通常低身長である。小児期または青年期に肥満になる可能性がある。 IQスコアは平均35~50であるが、発達の転帰は相当に様々である。EP300バリアントのRSTSの一部の方は知能正常である。その他の症状として、眼科疾患、難聴、呼吸困難、先天性心疾患、腎奇形、停留精巣、摂食障害、繰り返す感染症や重度の便秘がある。
診断・検査
RSTSの診断は発端者の特徴的な臨床所見によって診断する。臨床的所見が決定的でなければ、CREBBPまたはEP300 のヘテロ接合の病原性バリアントの同定による。
臨床的マネジメント
症状に対する治療:
早期教育プログラム、特殊教育、発達障害児に対応した職能訓練および行動異常の専門家、心理士、サポートグループ他の機関を家族に紹介; 眼科的異常、難聴、無呼吸、心疾患、腎奇形、停留精巣、歯の異常に対する標準治療;胃食道逆流症および便秘に対する積極的治療、著しく偏位した親指や重複母趾に対する外科的治療
サーベイランス :
特に生後1年間は成長および食事の経過観察、年1度の視力・聴力の検査、定期的な心臓、腎臓、歯の観察
妊娠に関する管理:
妊娠高血圧腎症や胎盤異常が報告されている。
遺伝カウンセリング
RSTSは常染色体顕性遺伝形式である。RSTS は典型的に、家族内の新生病原性バリアントとして発症する。ほとんどの患者が孤発例である (家族内で唯一、罹患している)。ほとんどの症例で、RSTS患者の両親は罹患していない。両親が臨床的に非罹患でも、体細胞または生殖細胞系列のモザイクによりヘテロ接合体をもつ片親の症状は軽度であり、同胞がRSTSである可能性はある。同胞の経験的再発リスクは1%以下である。RSTS患者が、子どもを産むことは稀であるが、理論的罹患率は50%である。家系内の患者で病原性バリアントが同定された場合、リスクの高い妊娠に対する出生前診断は可能である。
診断
臨床診断
RSTSの診断は以下の特徴的臨床所見や神経画像や家族歴があれば疑うべきである。
大症状
- 顔貌上の特徴(図1参照)
- 眼瞼裂斜下
- 外鼻孔より下方にのびた鼻柱を伴うかぎ鼻
- 高口蓋
- しかめっつら笑顔
- 切歯結節 (歯の舌側の副歯様構造物)永久歯列の上顎切歯に、しばしば生じる

図1 RSTSの顔貌上の特徴。アーチ状の眉、眼瞼裂斜下、鼻翼より下方に伸びた鼻柱、しかめっつら笑顔
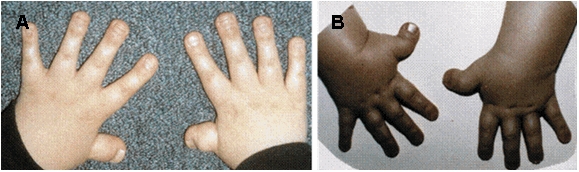
図2A 幅広の指節骨
図2B 幅広く橈側偏位した親指
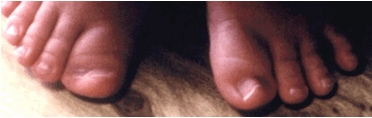
図3 幅広く、部分的に重なる親指
その他の症状(図2、3)
- 母指趾は幅広く、しばしば偏位している。
- 末節骨も幅広いことがある。
- 基節骨も異常な形態をしていることがある。手足のレントゲン写真は、異常を認めるが、必ずしも診断には必須ではない。
- 男児ではほとんど全例に停留精巣を認める。
- 尿路奇形がしばしば見られる。
- さまざまなタイプの先天性心疾患がRSTS患者の約1/3に見られる。
成長
- 胎児期における成長は正常であるが、生後数カ月で身長、体重および頭囲のパーセンタイルが急速に低下する。成人期では通常低身長である。
- 生後数か月より小頭症を呈し、成人期につれて典型的になる。
- 小児期または青年期に肥満になる可能性がある。
知的障害 平均IQスコアは35~50の範囲である。しかし、発達の転帰は、相当に様々である。一部のEP300バリアントのRSTSは知能正常である。
神経学的画像所見 脳画像において脳梁の異形成や形成不全を認める(73.6%)
家族歴 RSTSは典型的には新生病原性バリアントによって発症し、ほとんどの発端者は孤発例である(家系内に単独)。稀に常染色体顕性遺伝様式に一致する家族例がある(多世代にわたる罹患した男性・女性患者)
診断
RSTSの診断は前述の示唆した所見による。
臨床的所見が決定的でない場合、下記の分子遺伝学手検査の一つで同定できれば診断を確定できる(Table1)。
- CREBBPまたはEP300 のヘテロ接合の病原性(または病原性の可能性が高い)バリアント(罹患者の65-70%)[Fergelot et al 2016, Pérez-Grijalba et al 2019]
- 16p13.3 (CREBBP) oまたは 22q13.2 (EP300) 染色体のヘテロ欠失(罹患者の10%)[Negri et al 2015, Pérez-Grijalba et al 2019]
備考
(1)ACP/AMPバリアント解釈ガイドラインでは、臨床では「病原性バリアント」と「病原性バリアントの可能性が高い」という用語は同義であり、どちらも診断とみなされ、臨床的判断の決定として考慮できる [Richards et al 2015]。Gene Reviewにおける「病原性バリアント」への言及は、病原性バリアントの可能性が高いものを含む。(2)意義が不明なヘテロ接合性バリアントが同定されたとしても、診断が確定するわけでも、除外されるわけでもない。
分子遺伝学的解析は、症状によって、ターゲット遺伝子(単一遺伝子、多遺伝子パネル、マイクロアレイ染色体検査)検査や包括的ゲノム検査(エクソームシーケンシング、エクソームアレイ、ゲノムシーケンシング)を実施する。
ターゲット遺伝子検査は、臨床家がどの遺伝子を含むかを決定するが、包括的ゲノム検査は、その必要はない。RSTSの症状は幅広く、臨床的症状が特有であれば、ターゲット遺伝子検査(オプション1参照)を用いて診断し、低身長かつ/または知的障害などの多くの遺伝性疾患と鑑別が困難な表現型であればゲノム検査(オプション2参照)によって診断する。
オプション1
表現型および検査所見がRSTSの診断を示唆する場合、分子遺伝学的検査アプローチには、単一遺伝子検査または複数遺伝子パネルの使用が含まれる。染色体マイクロアレイ解析は、いくつかの状況で有用な場合がある。
- 症状を説明しえない限られた意義不明バリアント(VUS)や病原性バリアントなどの限られた遺伝子同定では、CREBBP、EP300及び他の関連遺伝子(参照 鑑別診断)を含む多重遺伝子パネルが最も合理的な費用で疾患の遺伝的原因を特定する可能性が最も高いであろう。
注:(1)パネルに含まれる遺伝子と、各遺伝子に使用される検査の診断感度は、検査室によって異なり、時間の経過とともに変化する可能性がある。(2)一部の複数遺伝子パネルには、このGene Reviewで説明されている疾患とは関連しない遺伝子が含まれている場合がある。(3)一部の検査室では、パネルオプションには、検査室で設計されたカスタムパネルおよび/または臨床医によって規定された遺伝子を含む表現型に重点をおいたカスタムエクソーム解析が含まれる場合がある。(4)パネルで使用される方法には、配列解析、欠失/重複解析、および/または他の非配列ベースの検査が含まれる場合がある。この疾患については、欠失/重複解析も含む複数遺伝子パネルが推奨される(表1を参照)。
- 染色体マイクロアレイ分析(CMA)は、オリゴヌクレオチドまたはSNPアレイを使用して、配列解析では検出できないゲノム全体での大きな欠失/重複(CREBBPおよびEP300を含む)を検出する。
注:(1)CREBBP病原性バリアントにおいて有意な割合で大きな欠失であるため、RSTSの診断を事前に考慮せずに実施されたCMAによってRSTSを診断する可能性がある。(2)CREBBPを含む隣接した遺伝子欠失とRSTSとの部分的な表現型の重複を呈するアレル疾患が報告されている(遺伝的関連疾患を参照)。
オプション2
包括的ゲノム検査では、臨床医がどの遺伝子が関与しているかを判断する必要はない。エクソームシーケンスが最も一般的に使用される。ゲノム配列解析も可能。報告されているCREBBP、 EP300の大部分の病原性バリアントは翻訳領域にありエクソームシークエンシングで同定される。
表1 ルビンスタイン・テイビ症候群で実施される分子遺伝学的検査
| 遺伝子1、2 | 病原性バリアントがRSTSに関与する割合 | 病原性バリアント3を同定できる割合 | |
|---|---|---|---|
| シーケンス解析4 | 遺伝子ターゲット欠失/重複解析5 | ||
| CREBBP | 55~60%6 | ~88% | ~12%7 |
| EP300 | 8~10%8 | ~93% | ~7% 9 |
| 不明10 | ~30% | NA | |
検査の実施に関してはGeneTests Laboratory Directoryを参照のこと。GeneReviewsは、分子遺伝学的検査について、その検査が米国CLIAの承認を受けた研究機関もしくは米国以外の臨床研究機関によってGeneTests Laboratory Directoryに掲載されている場合に限り、臨床的に実施可能としている。 GeneTestsは研究機関から提出された情報の検証や、研究機関の承認状態もしくは実施結果の保証は行わない。情報を検証するためには、医師は直接それぞれの研究機関と連絡をとる必要がある。
- 遺伝子リスト(アルファベット順)
- 染色体座位とたんぱくの遺伝子とデータベース 表A 参照
- 同定された遺伝子ヴァリアンの分子遺伝学的情報を参照
- シーケンス解析では benign、 likely benign、 of uncertain significance、 likely pathogenic、 pathogenic バリアントが検出される。バリアントには微細遺伝子内欠失/挿入やミスセンスバリアント、ナンセンスバリアント、スプライス部位バリアントを含む。通常、エクソンまたは遺伝子全体の欠失/重複は検出できない。
- 欠失/重複を標的とした遺伝子解析は遺伝子内欠失または重複が検出可能である。定量PCR法、長距離 PCR、MLPA法、単一エクソンの欠失・重複検出のために設計されたマイクロアレイなどが用いられる標的遺伝子の欠失・重複は単一エクソンから遺伝子全体までの欠失を検出するが、広範囲の欠失の断端または/かつ隣接遺伝子の欠失の検出は難しいだろう(Genetically Related (Allelic) Disorders参照)エクソームやゲノムのシークエンシングでは、断端の同定や深度を用いて欠失/重複を検出できる可能性があるが、感度は標的遺伝子の欠失/重複解析よりも低くなるであろう。
- Pérez-Grijalba et al [2019];Human Gene Mutation Databaseプロフェッショナルから得られたデータもある。[Stenson et al 2020]
- Spena et al [2015], Cross et al [2020]
- Negri et al [2015], Fergelot et al [2016]
- Fergelot et al [2016], Cohen et al [2020]
- RSTSの~30%は他の遺伝子の病原性バリアントによって生じる可能性がある。[Fergelot et al 2
臨床的特徴
自然経過
ルビンスタイン・テイビ症候群(RSTS)は、低身長、様々な構造異常、特徴的な顔貌、幅広な第1指指趾およびあらゆる程度の知的障害を特徴とする多系統疾患である。最も共通した頭蓋顔面の特徴は、小頭症、高度なアーチ状の眉毛、眼瞼裂斜下、凸状の鼻隆起、低く垂れ下がった鼻柱、しかめっつらの笑みである。親指と第一趾は幅広く、しばしば偏位している[Hennekam et al 1990, Stevens et al 1990]。
RSTSは、印象的な顔貌と特徴的な手足の所見のため、出生時または乳児期に認識されることが多い。幼少期の問題としては、呼吸困難、摂食障害、体重増加不良、感染症の再発、重度の便秘などがある。
現在までに少なくとも600人において、CREBBPまたはEP300の病原性バリアントが同定されている[Fergelot et al 2016, Pérez-Grijalba et al 2019, Cross et al 2020, Douzgou et al 2022]。この表現型の特徴に関する以下の記述は、これらの報告にもとづいている。
表2 ルビンスタイン・テイビ症候群の主な症状
| 症状 | 割合 | コメント |
|---|---|---|
| 成長障害 | 73% | |
| 眼 | 80% | |
| 難聴 | 30% | ほとんどが伝音性、感音性の場合もある。 |
| 呼吸器疾患 | Common | 感染症、誤嚥 |
| 心臓疾患 | 33% | |
| 泌尿生殖器症状 | 27% | 停留精巣が最も多い(罹患男性の78-100%) |
| 消化器系症状 | 88% | 摂食障害、便秘 |
| 骨格異常 | Common | 側弯20%, 第1指趾異常92% |
| 神経学的所見 | 21% | |
| 歯科所見 | 73% | 切歯結節, エナメル質低形成 |
| 皮膚所見 | 24% | ケロイド、毛母腫 |
| 繰り返す感染症 | 17% | 主に呼吸器 |
| 腫瘍 | 30% | 良性・悪性 |
| 発達遅滞 | 98% | |
| 行動異常 | 41% 自閉症/自閉的特徴, 27%-64% 不安症 | |
| 脳MRI異常 | 74% | 様々な所見 |
Wiley et al [2003], Schorry et al [2008], Stevens et al [2011], Milani et al [2015], Fergelot et al [2016], Boot et al [2018], Douzgou et al [2022]
成長 出生前の成長は通常正常であるが、生後1年内に成長障害を生じる。通常、思春期には成長スパートは認めない。EP300病原性バリアントをもつ患児では、小頭症と成長障害の発生率が高く、これはおそらく妊娠高血圧腎症の発生率上昇と関連している。BMIは21歳の男性では正常だが、女性では増加している。多くの成人が肥満である [ Stevenset al 2011 ]。成人男性の平均身長は162.6cm、成人女性の平均身長は151.0cm[ Beets et al 2014 ]。Beets et al [2014]は、RSTSの成長曲線を公開している。
眼:斜視、屈折異常、眼瞼下垂、鼻涙管閉塞、白内障、コロボーマ、眼振、緑内障、角膜異常が見られる。
難聴:再発または難治性の中耳疾患は、伝音難聴を引き起こす可能性がある。感音性難聴もある。
呼吸器:閉塞性無呼吸はしばしば考慮すべき問題となる。これは狭い口蓋、小顎、筋緊張低下、肥満、喉頭壁の易虚脱性等が原因で生じる。挿管と麻酔の合併症の発生が報告されている。誤嚥、喘息、および再発性上気道感染症も生じる可能性がある。[Bradford et al 2022].
心臓:約1/3にさまざまな先天性心疾患を合併する(例 心房中隔欠損症、心室中隔欠損症、動脈管開存症、大動脈縮窄症、肺狭窄症、二尖大動脈弁、偽性動脈症、大動脈弁狭窄症、血管輪、伝導障害)。
泌尿生殖器:水腎症や重複尿管などの腎臓の異常は非常に一般的である。その他の泌尿生殖器の合併症には、尿道下裂、膀胱尿管逆流、腎結石症および尿路感染症が含まれる。ほとんどすべての男児は停留精巣を有する。
消化器:摂食障害、胃食道逆流症、便秘が一般的。胆汁性嘔吐、再発性腹痛、便の通過障害または血便がある場合は、回転異常を疑うべきである[Stevens 2015]。
整形外科:著しく偏位した母指と重複した母趾に加えて、整形外科的には、膝蓋骨脱臼、関節弛緩、脊椎わん曲、レッグペルテス病、大腿骨頭すべり症、頸椎の異常などがある。
神経:キアリ奇形、脊髄空洞症、歯突起骨および頸髄圧迫を含む頭蓋脊髄および後頭蓋窩異常が時折。報告されている [ Marzuillo et al2013 ]。脊髄係留または脂肪腫もある。けいれんまたは異常EEG所見が認めることがある。
歯:歯の叢生、不正咬合、複数の齲歯、乏歯、過剰歯、魔歯、永久歯の上顎切歯
皮膚:ほんのわずかな外傷でもケロイドを生じることがある。石灰化上皮腫が報告されている。
繰り返す感染症:中耳炎、肺炎、およびその他の呼吸器感染症などが報告されている。体液性または細胞性免疫不全の報告あり。
腫瘍:神経芽細胞腫、胎児型横紋筋肉腫、髄芽腫、悪性血液疾患など様々な腫瘍の合併報告がある。最近のオランダ人のRSTSを対象とした研究では、悪性腫瘍のリスクの増加は確認されていない。但し、髄膜腫と毛母腫の有意な発生率の上昇を認めた[ Boot et al 2018 ]。現在、悪性腫瘍に対する40歳以前の追加サーベイランスの推奨はない。
内分泌:持続性高インスリン性低血糖は、主にEP300の病原性バリアントを有するRSTS患児の小数名で報告されている[Welters et al 2019]
思春期:思春期および性的発達は正常である。
発達と知能:RSTSの小児は通常、発達の遅れを認める。ある研究では、独歩獲得は平均30カ月、始語獲得は平均25カ月、トイレトレーニングは平均62カ月であった。言語発達の遅れは90%の児で見られ、ほとんど言葉を話さない者も一部いる。Waite et al [2016]は、言語的および視覚空間的作業記憶の欠如を指摘している。ある研究では、平均IQ 51、他の研究では36であった。IQスコアは25~79の範囲である。動作性IQは言語性IQより高くなることが多い。EP300-RSTSの一部の人は知能正常であった[Fergelot et al 2016]。RSTSの成人を対象とした研究では、家族は、社会的相互作用の減少、限られた会話、持久力やスタミナと移動の低化など、時間の経過とともに能力の32%低下を報告した[Stevens et al 2011]。
行動:衝動性、注意散漫性、気分の不安定性、および常同行動が頻繁に観察される[Verhoeven et al 2010 ]。その他の行動異常には、注意欠陥、多動性、自傷行為、攻撃的な行動などがある。RSTSの成人の約62%が自閉症様の行動、3分の1の人が理由のない恐れや不安を有してした[Stevens et al 2011]。同じものや質問の繰り返しにこだわりがあるかもしれない[Waite et al 2015]。Crawford et al [2017]は、パニック発作、広場恐怖症、強迫性障害のレベルが高いことを指摘した。
脳MRI所見:脳画像上の最も一般的な特徴は、脳梁異形成または形成不全(73.6%)である。それらは、小脳虫部軽度形成不全や脳室周囲後方白質病変やその他異常所見を伴う・伴わないこともある。その他の稀な所見には、キアリ奇形、ダンディ・ウォーカー奇形および未発達な下垂体がある。
予後:RSTS患者の90%以上が成人期まで生存する[Milani et al 2015]。
RSTSの寿命が異常かどうかは不明である。67歳の生存報告もあり[Stevens et al 2011]、成人期までの生存が可能であることを示している。障害を持つ成人の多くは高度な遺伝子検査を受けていないため、成人のRSTSは認識されず、診断に至っていない可能性が高い。
遺伝子型と臨床型の関連
EP300の病原性バリアントは、 CREBBP -RSTSに似た表現型を生じる。ただし、下方にのびた鼻中隔を除いて、 EP300 -RSTSの顔の特徴はあまり目立たない。親指と母趾は幅広いが、偏位した母指は非常にまれである。知的障害はさまざまで、通常はそれほど重度ではなく、時には正常である [ Fergelot et al 2016 ]。
遺伝子型と臨床型の関連
CREBBPとEP300における病原性バリアントの種類と位置は、特定の身体的特徴、先天異常、認知、行動とは相関しない。例外は、CREBBPとEP300の両方のエクソン30末端とエクソン31の先頭のミスセンスバリアントである。この表現型はRSTSとは異なり、Menke-Hennekam症候群として知られている。
EP300 多くのバリアントを検討した研究では、全体的な表現型の重症度と、病原性バリアントの型や、HATドメインやエクソン31に関連する位置に相関は認められなかった。遺伝子型と知的障害や重症度や主要臓器の重症度にも同様に相関は認められなかった [Cohen et al 2020]。
CREBBP現在までに多くのRSTS患者においてCREBBPを含む様々な大きさの欠失が報告されている。Stefら[2007]とPérez-Grijalbaら[2019]は、CREBBP欠失の大きさによる表現型の違いを認めなかった。Rusconiら[2015]は、CREBBPの単一エクソンから全遺伝子およびフランキング領域欠失を有する14人について報告した。CREBBP含む欠失の場合、ミスセンスの病原性バリアントよりも必ずしも重症型ではないことを指摘した。
Spena et al [2015]は、ヒストンアセチルトランスフェラーゼドメイン外の病原性バリアントは軽度な表現型と関連する可能性を示唆した。Pérez-Grijalbaら[2019]は、病原性バリアントのタイプ、位置、HATドメイン含有と重症度との相関は認められなかったと報告した[Gervasini et al 2007、Chiang et al 2009 ]。
Gervasini et al [2007] and Schorry et al [2008]によってモザイク微小欠失が報告されている。モザイク微小欠失を有する患者は非モザイク欠失の患者より軽い重症度の表現型を呈する傾向であった。
有病率
Hennekam et al [1990b]は、オランダのRSTSの出生率が1:100,000から1:125,000であると報告している。
遺伝学的に関連のある疾患(同一アレル疾患)
遺伝子内CREBBPおよびEP300病原性バリアント 生殖細胞系のCREBBPおよびEP300の病原性バリアントは、Menke-Hennekam症候群(OMIM 618332および618333)に関連している。これは、CREBBPのエクソン30の最後の部分またはエクソン31の最初の部分またはEP300の相同領域のミスセンスバリアントによって生じる。Menke-Hennekam症候群では、RSTSに特徴的な顔貌や広い/偏位した母指や母趾を共有していない。顔の特徴には、眼瞼下垂、眼角隔離、短く上斜めの眼瞼裂、平坦な鼻隆起、短い鼻、上向き鼻孔、短い鼻中隔、および長い人中を呈する。その他の特徴には、低身長、知的障害、小頭、摂食困難、けいれん、自閉症行動、およびその他のさまざまな所見がある[ Menke et al 2016、Menke et al 2018、Banka et al2019 ]。
CREBBPの微細欠失/重複 CREBBP遺伝子を包含する16p13.3の微小重複 [Demeer et al 2013]は、(RSTSとは類似しない)特徴的な顔貌、心臓および腎臓疾患、口蓋裂、脳梁の形成異常を特徴とする [Kang et al 2023]。16p13.3欠失(重症RSTSとしても知られる)には、CREBBP遺伝子のほか、様々な隣接する他の遺伝子が含まれる。臨床的特徴としては、成長障害、脳の先天異常、けいれん、難治性感染症などがある [Bartsch et al 2005]。
鑑別診断
特徴的顔貌および手足の異常からRSTSの診断は容易である。
幅広く、偏位した母指趾はFGFR関連の頭蓋骨癒合症(Pfeiffer症候群およびApert症候群)、Saethre-Chotzen症候群、Greig頭蓋多合指症候群においてもみられる。頭蓋骨癒合の有無や顔貌の特徴からこれらの疾患と鑑別する(表3参照表3 ルビンシュタイン・テイビ症候群(RSTS)の鑑別診断における他の関連遺伝子
| 遺伝子 | 鑑別診断する疾患 | 遺伝様式 | 鑑別診断する疾患の臨床的特徴 | |
|---|---|---|---|---|
| オーバーラップとRSTS | RSTSとの区別 | |||
| FGFR1 FGFR2 FGFR3 |
ファイファー症候群およびアペール症候群(FGFR関連の頭蓋骨癒合症症候群を参照) | AD | 広い/偏位した母指と母趾 |
|
| GLI31 | 典型的なGreigcephalopolysyndactyly症候群(GCPS) | AD | 広い/偏位した母指と母趾 |
|
| GPC4 | Keipert syndrome (OMIM 301026) | XL | 幅広の母指と母趾 |
|
| HOXD13 | 短指症タイプD (OMIM 113200) |
AD | 片側または両側親指の末節骨の短縮 |
|
| SRCAP 2 | Floating-Harbor 症候群 | AD |
|
|
| TWIST1 | 古典的なSaethre-Chotzen症候群 | AD | 広い/偏位した母指と母趾 |
|
| NIPBL SMC1A SMC3 RAD21 HDAC8 BRD4 | Cornelia de Lange syndrome | AD, XL |
低身長、顔貌、知的障害 | 四肢欠損 |
| KMT2A | Wiedemann-Steiner syndrome | AD | 低身長、顔貌、知的障害 | 肘部多毛症、典型的な手足の所見の欠如 |
AD =常染色体優性遺伝; DD =発達遅延; ID =知的障害; MOI=遺伝様式;OFC =後頭前頭周囲
RSTS=ルビンスタイン・テイビ症候群
- Greigcephalopolysyndactyly症候群は、GLI3 のヘテロ接合性の病原性バリアントまたは GLI3 を含む染色体 7p14.1 の欠失と関連している。
- Floating-Harbor syndrome症候群は、CREB 結合タンパク質のコアクチベーターとして機能する SNF2 関連クロマチンリモデリング因子をコードする SRCAP の病原性バリアントによって生じる。このため RSTS との表現型とオーバーラップすると考えられる。
臨床的マネジメント
ルビンスタイン・テイビ症候群(RSTS)の臨床診療ガイドラインは、Wileyら[2003]によって報告されている。
最初の診断に続いて行う評価
RSTS と診断された人の疾患の程度とニーズを確立するために、表 4 にまとめられている評価 (診断につながった評価の一部として実行されていない場合) が推奨される。
表4 ルビンシュタイン・テイビ症候群の個人における初期診断後の推奨される評価
| 臓器/関連 | 評価 | コメント |
|---|---|---|
| 体格 | 成長の測定 | RSTS成長チャートに数値をプロットする。 |
| 神経 |
|
|
| 発達 | 集学的な発達および/または神経心理学的評価 | ・運動、適応、認知、言語評価を含める ・早期介入/特別教育の評価 |
| 神経行動精神 | 神経行動学的評価 | 12か月以上の場合:睡眠障害、ADHD、不安、および/またはASDを示唆する所見を含む事項のスクリーニング |
| 筋骨格 | 整形外科 / 理学療法とリハビリテーション / PT と OT の評価 |
|
| 胃腸・栄養 | 消化器科 / 栄養学 / 摂食チームの評価 |
|
| 眼科 | 眼科検査 | 斜視、屈折異常、眼瞼下垂、鼻涙管閉塞、白内障、コロボーマ、眼振、緑内障、および角膜異常について評価する。必要時、専門医療や低視力サービスへの紹介 |
| 聴力 | 聴力評価 |
|
| 心血管 | 心臓の評価 |
|
| 呼吸器 | 肺の評価 | いびき、特定の睡眠姿勢、夜間覚醒、および日中の過度の眠気がある場合、睡眠ポリグラフ検査で閉塞性睡眠時無呼吸症候群の評価を行う。 |
| 泌尿生殖器 | 腎臓・泌尿器の評価 |
|
| 歯科/歯科矯正 | 歯科および歯列矯正の評価 | 口蓋、歯の数と位置、前歯の中心結節、齲歯、歯周病の評価 |
| 内分泌 | 内分泌の評価 | 神経過敏、筋力低下、発作などの低血糖症状がある場合は、高インスリン血症を評価する。 |
| 遺伝カウンセリング | 遺伝専門家1 | 家系図を入手し、RSTSや家族構成の復元、遺伝様式、および罹患者とその影響を受けた人々とその家族に知らせ、医療および個人的な意思決定を支援する。 |
| 家族支援/資源 | 臨床医、幅広いケアチーム、家族支援組織による | 必要な資源を判断するため、家族と社会構造を評価する。
|
Wiley et al [2003]
ADHD = 注意欠陥・多動性障害、ADL = 日常生活動作、ASD = 自閉症スペクトラム障害、GI = 胃腸、MOI = 遺伝形式、OT = 作業療法、PT = 理学療法、RSTS = ルビンスタイン・テイビ症候群、VCUG = 排尿時膀胱尿道造影検査
- 医療遺伝学者、認定遺伝カウンセラー、認定専門遺伝看護
治療
RSTS には治療法はない。
生活の質を改善し、機能を最大限に高め、合併症を軽減するための支持療法が推奨される。関連分野の専門家による多領域にわたる治療が理想的である。(表 5 を参照)。
表5 ルビンシュタイン・テイビ症候群における症状の治療
| 症状/関連 | 治療 | 考慮事項/ その他 |
|---|---|---|
| 発達遅滞/知的障害/行動関連 | 発達遅滞/知的障害管理を参照 | |
| 体重増加不良 /発育不全 |
|
嚥下障害の臨床的兆候または症状がある場合、臨床的な摂食評価および/または嚥下造影検査を検討する。 |
| けいれん | 経験豊富な神経科医による抗発作薬の標準治療 |
|
| 筋・骨 | 整形外科 / 理学療法とリハビリテーション /拘縮や転倒を防ぐためのストレッチを含む理学療法と作業療法 |
|
| 消化管 | 消化器医/栄養士による標準治療 |
|
| 眼の症状 | 眼科医による標準治療 | 屈折異常、斜視 |
| 眼科専門医 | 複雑な所見(例:白内障、網膜ジストロフィー、緑内障) | |
| ロービジョンサービス |
|
|
| 難聴 | 聴覚訓練士による標準治療 | 早期介入または学区によるコミュニティ聴覚サービス |
| 心臓の異常 | 心臓病専門医による標準治療 |
|
| 耳鼻科 | 閉塞性睡眠時無呼吸症/再発性中耳炎の標準治療 | 臨床的に適応がある場合は、睡眠ポリグラム、CPAP、扁桃腺/アデノイドの除去、鼓室チューブ |
| 泌尿生殖器 | 腎臓医/泌尿器科医による標準治療 |
|
| 歯の異常 | 歯科医および/または歯科矯正医による標準治療 | 咬み合わせ、閉口や齲歯の原因となった前歯の中心結節の治療 |
| 皮膚 | ケロイド/毛母腫のモニター |
|
| 家族/地域 |
|
在宅介護サポートの必要性のための継続的な評価 アダプティブスポーツやスペシャルオリンピックス参加の検討 |
ASM = 抗てんかん薬、CPAP = 持続的陽圧呼吸、OT = 作業療法、PT = 理学療法
- 親や保護者に一般的な発作の症状について教育することは適切である。てんかんと診断された子供に対する非医学的介入と対処戦略に関する情報については、Epilepsy Foundation Toolbox を参照。
発達遅滞・知的障害 医療管理問題
以下の情報は、米国における発達遅滞/知的障害のある人に対する一般的な管理推奨事項を示している。標準的な推奨事項は国によって様々に異なることがある。
0-3歳 作業療法、理学療法、言語療法、摂食療法のほか、乳児メンタルヘルス サービス、特別教育者、感覚障害の専門家を利用するには、早期介入プログラムへの紹介を推奨する。米国では、早期介入は連邦政府が資金提供するプログラムであり、すべての州で利用可能で、個人の治療ニーズを対象とした在宅サービスを提供する。
3-5歳 米国では、地元の公立学区を通じた発達プレスクールが推奨される。配置前に、必要なサービスと療育法を決定するための評価が行われ、運動、言語、社会、または認知の遅れにもとづいて、個別教育計画 (IEP) が作成される。早期介入プログラムは通常、この移行を支援する。発達プレスクールはセンターベースである。医学的に不安定で通学できない子供には、在宅サービスが提供される。
全年令 両親の生活の質を最大限に高めるための支援に、適切な地域、州、教育機関(米国)との関わりを確保し、発達小児科医との相談が推奨される。考慮すべきいくつかの問題点:
- IEP (個別支援プラン)サービス
- IEP は、資格のある子どもたちに、特別に作成された指導と関連したサービスを提供する。
- IEP サービスは毎年見直され、変更が必要かどうか判断される。
- 特別教育法では、IEP に参加する児童は、いつでもどこでも、学校で可能な限り制限の少ない環境におかれ、また一般教育に参加することを義務付けている。
- 視覚と聴覚のコンサルタントは、学習教材へのアクセスの支援するために、子どもの IEP チームの一員とすべきである。
- 理学療法、作業療法、言語サービスは、その必要性が児童の教科学習へのアクセスに達するまで、IEP の中で提供される。それ以上の場合は、個人のニーズによって民間の支援療法が考慮される。発達小児科医は、療育の種類など具体的な推奨を行う。
- 子どもが 10 代に入ると、移行計画を話し合い、IEP に組み込む必要がある。公立学校区は IEP のサービスを受けている人たちに、21 歳までサービスを提供する必要がある。
- A504プラン(セクション 504: 障害に基づく差別を禁止する米国連邦法)は、教室前方の座席、支援技術機器、教室の筆記者、授業間の時間延長、課題の変更、テキストの拡大などの配慮や変更を検討する。
- 発達障害局(DDA)への登録の推奨。DDAは、資格のある個人にサービスと支援を提供する米国の公的機関である。資格は州によって異なるが、通常は診断および/または関連する認知/適応障害によって判断される。
- 収入や資産が限られている家族は、障害のある子供のための補足的保障所得(SSI)の資格を得ることが可能。
運動機能障害
粗大運動機能障害
- 可動性を最大限に高め、後期発症する整形外科的合併症(拘縮、脊柱側弯症、股関節脱臼など)のリスクを軽減するために、理学療法が推奨される。
- •必要に応じて、耐久性のある医療機器や体位変換機器(車椅子、歩行器、バスチェア、矯正器具、適応型ベビーカーなど)の使用を検討。
- •筋緊張亢進やジストニアなどの筋緊張異常については、バクロフェン、チザニジン、ボトックス®、抗パーキンソン病薬、または整形外科治療の管理を支援するために、適切な専門家の関与を検討する。
微細運動機能障害 食事、身だしなみ、着替え、筆記などの適応機能に影響する微細運動能力の障害には作業療法が推奨される。
口腔運動機能障害 診察ごとに評価する必要があり、哺乳中の窒息/えずき、体重増加不良、頻繁な呼吸器疾患、または他に説明のつかない哺乳困難について、摂食評価および/また造影検査を行う必要がある。子供が安全に経口哺乳できる場合は、協調性または感覚関連の哺乳問題を改善するために、摂食指導(通常は作業療法士または言語療法士による)が推奨される。安全のために、栄養を濃くしたり冷やしたりする。哺乳機能障害が重度の場合は、経鼻チューブまたは胃瘻チューブが必要になる場合がある。
コミュニケーションの問題。表現言語に困難がある人に対しては、代替コミュニケーション手段(例:補助代替コミュニケーション [AAC])の評価を検討する。AAC 評価は、この分野の専門知識を持つ言語聴覚療法士が行うことができる。評価では、認知能力と感覚障害を考慮して、最も適切なコミュニケーション形式を決定する。AAC デバイスは、絵カード交換コミュニケーションなどのローテクなものから、音声生成デバイスなどのハイテクなものまで多様である。一般に信じられていることとは異なり、AAC デバイスは発話の言語発達を妨げるものではなく、むしろ最適な発話と言語の発達をサポートするものである。
神経行動学・精神医学上の問題
子どもは、応用行動分析 (ABA) などの自閉症スペクトラム障害の治療に使用される介入の対象となり、その恩恵を受けることができる。ABA 療法は、個々の子どもの行動、社会性、適応の長所と短所を対象としており、通常は認定行動分析士と 1 対 1 で実施される。
発達小児科医との相談は、適切な行動管理戦略について親を指導したり、必要に応じて注意欠陥多動性障害の治療薬などの処方薬を提供したりするために役立つ。
重度の攻撃的または破壊的な行動に関する問題は、小児精神科医が対処することが可能。
サーベイランス
既存の症状、支持療法に対する個々の反応、および新たな症状の出現を監視するために、表 6 にまとめた評価が推奨される
表6 ルビンシュタイン・テイビ症候群に推奨されるサーベイランス
| 臓器/関連 | 評価 | 頻度 |
|---|---|---|
| 成長 | RSTS成長曲線を用いて体重と成長を観察記録。 |
|
| 神経 | 臨床的な発作のある場合、モニター | 診察ごと |
| 発作、緊張の変化、運動障害などの新たな症状の評価 | 診察ごと | |
| 発達 | 発達の進捗と教育ニーズをモニター | 診察ごと |
| 神経行動/精神 | 不安、ADHD、ASD、攻撃性、自傷行為の評価 | 診察ごと |
| 筋骨格 |
|
診察ごと |
| 栄養 |
|
診察ごと |
| 消化器系 | 便秘のモニター | 診察ごと |
| 呼吸器系 | 誤嚥、呼吸不全、睡眠時無呼吸の兆候のモニター | 診察ごと |
| 眼の症状 | 眼科的評価 | 毎年または必要に応じて |
| ロービジョンサービス | 臨床医ごと | |
| 難聴 | 聴力評価 | 毎年(再発性中耳炎がある場合はより頻回) |
| 心血管系 | 心機能評価 | 循環器医による診断とその後の指示 |
| 生殖泌尿器系 | 腎臓、泌尿器の評価 | 診断後、診察ごとに症状をモニター。 |
| 歯の異常 | 歯科および歯列矯正の評価 | 1歳から。6か月ごとに、または歯科医/歯科矯正医により継続 |
| 内分泌系 | 低血糖の評価 | 診察ごと |
| 免疫系 |
|
診察ごと |
| 家族/地域 | 新たな問題(家族計画など)が生じた場合、ソーシャルワークによるサポート(緩和ケア、レスパイトケア、在宅看護、その他の地域資源など)、ケアの調整、または遺伝カウンセリングのフォローアップなど必要性の評価。 | 診察ごと |
ADHD = 注意欠陥・多動性障害、ASD = 自閉症スペクトラム障害、OT = 作業療法、PT = 理学療法、RSTS = ルビンスタイン・テイビ症候群
リスクのある親族の検査
リスクのある親族への検査に関する事柄等は「遺伝カウンセリング」の項を参照。
妊娠管理
EP300 -RSTSの胎児を有した母52人中12人とCREBBP -RSTS胎児を有した母 59人中2人に妊娠高血圧腎症を認めた [ Fergelotら、2016 ]。別の研究では、EP300関連RSTSの個人12人中4人が妊娠高血圧腎症または胎盤異常を合併しており、子宮胎盤灌流不全の報告が1例あった。羊水過多症は2例の妊娠で認められた[Cohen et al 2020]。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ルビンスタイン・テイビ症候群(RSTS)は常染色体顕性遺伝形式である。
家族構成員のリスク
発端者の両親
- ほとんどのRSTSは新生突然病原性バリアントである。家族のなかで唯一の罹患者である。
- まれに、CREBBPまたはEP300 病原性バリアントを有しRSTSと診断された人が、ヘテロ接合またはモザイクの親からの由来であることがある。
- 臨床的表現は多様であるため、正常な知能を持つ親がCREBBPまたはEP300病原性バリアントのヘテロ接合性である可能性はわずかながらある[Bartsch et al 2010、López et al 2016]。EP300はRSTS表現型軽症と関連しているため[López et al 2016]、一見無症状の親の場合、EP300病原性バリアント(すなわち、CREBBP病原性バリアントと比較して)では親のヘテロ接合性は臨床的に高い疑いがある[López et al 2016]。
- 親の体細胞および生殖細胞モザイクが報告されている[Chiang et al 2009、Bartsch et al 2010、Tajir et al 2013、Lin et al 2021]。
- 発端者で分子遺伝学的に診断されている場合、発端者の親について、RSTSに関連する身体所見の診察、および発端者で同定されたCREBBPまたはEP300 病原性バリアント分子遺伝学的検査を推奨する。
- 発端者はで同定された病原性バリアントが両親で検出できず、親の身元検査で生物学的母性と父性が確認された場合は、以下の可能性を考慮する必要がある。
- 発端者は新生の病原性バリアントを有する。
- 発端者は、生殖細胞系列(または体細胞系列と生殖細胞系列)モザイクの親から病原性バリアントを受け継いでいる[Chiang et al 2009、Bartsch et al 2010、Tajir et al 2013、Lin et al 2021]。*親の白血球DNAの検査では、体細胞モザイクのすべての例を検出できない可能性があり、生殖細胞のみに存在する病原性バリアントは検出されない。
*親が病原性バリアントを最初に発生した個人の場合、親はバリアントの体細胞モザイクを有し、軽度または最小限の軽微な症状の可能性である。[Chiang et al 2009、Bartsch et al 2010]。
- RSTSと診断された人の家族歴は、家系員の症状を認識できずに否定される場合がある。そのため、発端者の両親に対して発端者で同定された病原性バリアントのヘテロ接合性の分子遺伝学的検査が行われない限り、家族歴の陰性は明らかには確定はできない。
発端者の同胞
発端者の同胞に対するリスクは、発端者の両親の臨床的・遺伝的状態による
- 発端者の親が罹患している場合、および/または発端者で同定されたCREBBPまたはEP300 病原性バリアントを有している場合、同胞へ遺伝する可能性は50%。
- 発端者の有するCREBBPまたはEP300 病原性バリアントをいずれかの親の白血球DNAで検出できない場合、発端者は新規病原性バリアントを有している可能性か最も高いと説明づけられる。しかし、親の体細胞および/または生殖細胞のモザイクの可能性があるため、同胞への再発リスクは依然として一般集団のリスクよりも大きい。RSTSの親で体細胞および生殖細胞系列のモザイクが報告されている[ Chiang et al 2009、Bartsch et al 2010、Tajir et al2013 ]。
- 親が原因となる病原性バリアントの検査をうけておらず、かつ、明らかに無症状である場合、ヘテロ接合または体細胞および/または生殖細胞モザイクによる軽症の可能性があるため、同胞はRSTSの遺伝性の可能性がなお高いと推定される。同胞の経験的再発リスクは1%未満である。
発端者の子
RSTSの子供は、RSTS関連の病原性バリアントを引継ぐ可能性が50%ある [Hennekam et al 1989、Marion et al 1993、Petrij et al 2000、Bartsch et al2010 ]。
他の家族構成員
発端者の他の家族へのリスクは、発端者の両親の遺伝的状態による:親が病原性バリアントを有する場合、彼または彼女の家系員は罹患リスクの可能性がある。
関連する遺伝カウンセリング上の諸事項
早期診断・早期治療を目的としてリスクを有する血族に対して行う評価関連の情報については、「管理」の中の「リスクを有する血縁者の評価」の項を参照されたい。
家族計画
- 遺伝リスクの推定や出生前/着床前遺伝・診断の利用については、妊娠前に話し合うことが最適である。
- RSTSの子供をもうけた若年成人に遺伝カウンセリング(子孫への潜在的なリスクと生殖の選択の議論を含む)を提供することは適切である
DNAバンキング
DNAバンクは主に白血球から抽出したDNAを将来の使用のために保存しておくものである。検査法や遺伝子、遺伝子バリアントや疾患に対する知識が進展する可能性があり、罹患者のDNAの保存は検討する必要がある。
出生前検査ならびに着床前遺伝学的検査
罹患した家系員でRSTSの原因となる病原性バリアントが同定されると、リスクが高い妊娠の出生前検査と、RSTSの着床前遺伝子検査が可能になる。
先験的な低リスク妊娠。RSTSは通常、出生前超音波では診断されない。ただし、定期的な出生前超音波検査では、成長障害、羊水過多症、幅広の親指、脳の異常など、遺伝的リスクが高くない胎児において、RSTSの可能性がある所見を検出する可能性はある [ Van-Gils et al 2019 ]。
医療専門家間および家族内で、出生前検査について、見解の相違が生じる可能性がある。多くの医療機関では出生前検査を個人的意思決定とみなすが、これらの問題について議論することは有用であろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- National Library of Medicine Genetics Home Reference
Rubinstein-Taybi syndrome
- Understanding Rubinstein-Taybi syndrome: A Guide for Families and Professionalswww.Ucucedd.org
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表 A ルビンシュタイン・テイビ症候群:遺伝子とデータベース
| 遺伝子 | 染色体座位 | タンパク質 | 遺伝子座固有のデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| CREBBP | 16p13.3 | CREB結合タンパク質 | CREB Binding Protein (CREBBP) @ LOVD | CREBBP | CREBBP |
| EP300 | 22q13.2 | ヒストンアセチルトランスフェラーゼp300 | E1A binding protein p300 (EP300) @ LOVD | EP300 | EP300 |
データは、次の標準参照から編集されている:HGNCからの 遺伝子; OMIMからの 染色体座位遺伝子座; UniProtからのタンパク質。
表B ルビンシュタイン・テイビ症候群のOMIMエントリー| 180849 | RUBINSTEIN-TAYBI SYNDROME 1; RSTS1 |
| 600140 | CREB-BINDING PROTEIN; CREBBP |
| 602700 | E1A-BINDING PROTEIN, 300-KD; EP300 |
| 613684 | RUBINSTEIN-TAYBI SYNDROME 2; RSTS2 |
CREBB結合タンパク質(CREBBP)は一様に発現しており、多くの異なる転写因子の転写共活性化に関与している。内在性のヒストンアセチルトランスフェラーゼ(HAT)活性を有し、クロマチンリモデリングを介して転写複合体とのタンパク質相互作用を安定化する足場として機能する。CREBBPは、細胞増殖制御、細胞分化、アポトーシス、腫瘍抑制などの細胞経路に影響を与える多くの遺伝子の発現を調節している[ Negri et al2016 ]。CREBBPの生殖細胞系病原性バリアントは、短縮型またはアミノ酸置換のCREB結合タンパク質になる。HATドメインの病原性バリアントは転写活性化の重要な段階であるヒストンのアセチル化を抑制する。CREBBPは、多くのヒトの癌で制御する働きをする腫瘍抑制系であるp53もアセチル化する。
EP300は、アミノ酸レベルでCREBBPと63%の相同性を共有するp300転写コアクチベータータンパク質をコードする。HATとして、クロマチンリモデリングを介して転写を調節し、細胞の増殖と分化に重要な役割を果たす。病原性バリアントは、短縮したp300タンパク質または対立遺伝子発現の消失をもたらし、HAT活性を不活化する可能性がある。
疾患の発症機構 機能喪失(ハプロ不全)
更新履歴:
- GeneReview 著者: Malanie G Pepin, MS; Peter H Byers, MD
日本語訳者:古庄知己(信州大学医学部附属病院遺伝子診療部)
GeneReview 最終更新日: 2000.4.15. 日本語訳最終更新日: 2003.8.20. - Gene Review著者:Cathy A Stevens, MD
日本語訳者:古庄知己(信州大学医学部附属病院遺伝子診療部)、吉江幸司(信州大学医学部医学科)
Gene Review 最終更新日2004.9.13 日本語訳最終更新日2005.1.13 - Gene Review著者: Cathy A Stevens, MD
日本語訳者: 吉村祐実(翻訳ボランティア),小崎里華(国立成育医療研究センター病院遺伝診療科)
Gene Review 最終更新日: 2009.8.20. 日本語訳最終更新日: 2013.1.1. -
GeneReviews著者: Cathy A Stevens、 MD.
日本語訳者:小崎 里華 (国立成育医療研究センター 遺伝診療科)
GeneReviews最終更新日: 2019.8.22. 日本語訳最終更新日: 2021.5.18. - Gene Reviews著者: Cathy A Stevens、 MD
日本語訳者:小崎 里華 (国立成育医療研究センター 遺伝診療科)
GeneReviews最終更新日: 2023.11.9. 日本語訳最終更新日:2024.10.14.[in present]
| X POST |
![]()