ワイス・クルシュカ症候群
(Weiss-Kruszka Syndrome)
[Synonyms:ZNF462 異常症]
Gene Reviews著者: Paul Kruszka, MD, MPH.
日本語訳者: 沼部博直 (東京都立北療育医療センター)
GeneReviews最終更新日: 2019.10.31 日本語訳最終更新日: 2023.04.11.
要約
疾患の特徴
ワイス・クルシュカ症候群は前頭縫合隆起または頭蓋縫合早期癒合症,眼瞼下垂,非特異的形態変化,発達遅滞,自閉症状を特徴とする.脳の画像検査では脳梁の異常を認めることがある.発達遅滞は全般性であることも,運動発達遅滞であることも,言語発達遅滞であることもある.罹患者は耳の異常,摂食障害(胃瘻チューブの設置を要することもある),先天性心疾患を有することもある.臨床症状は同一家系内の患者であっても極めて多様性に富む.
診断・検査
ワイス・クルシュカ症候群は前頭縫合隆起または頭蓋縫合早期癒合症,眼瞼下垂,非特異的形態変化,発達遅滞,自閉症状を特徴とする.脳の画像検査では脳梁の異常を認めることがある.発達遅滞は全般性であることも,運動発達遅滞であることも,言語発達遅滞であることもある.罹患者は耳の異常,摂食障害(胃瘻チューブの設置を要することもある),先天性心疾患を有することもある.臨床症状は同一家系内の患者であっても極めて多様性に富む.
臨床的マネジメント
対処療法:
頭蓋縫合早期癒合症のある患者は頭蓋顔面外科チームや脳神経外科医に紹介; 摂食障害のある患者には摂食療法; 摂食障害の持続や嚥下障害のある患者には胃瘻チューブ造設.眼瞼下垂,発達遅滞,自閉症,難聴ならびに先天性心疾患には標準的治療を行う. サーベイランス: 幼児期から小児期早期にかけての定期検診時の頭囲と頭部形状の評価.診察毎の身体発育値の測定,栄養状態の評価,経口摂取の安全性の確認,神経発達状態と教育支援の必要性の評価.臨床的必要性に応じての眼科的ならびに耳科的評価.
遺伝カウンセリング
ワイス・クルシュカ症候群は常染色体顕性(優性)遺伝形式で遺伝する.ワイス・クルシュカ症候群患者の約95%は明らかに新生病的バリアントによる[親世代からの遺伝ではない].ワイス・クルシュカ症候群患者の子には50%の確率でZNF462遺伝子の病的バリアントが遺伝する.ZNF462遺伝子の病的バリアントを受け継いだ子は,家系内でも臨床的多様性があるため,患者より症状が強いことも弱いこともある.家系内のZNF462遺伝子の病的バリアントが同定されている場合,リスクの高い妊娠における出生前診断や着床前遺伝学的検査が可能である.
診断
ワイス・クルシュカ症候群の公式な診断基準は確立されていない.
臨床診断
下記の臨床的ならびに頭部MRI所見を有する場合にはワイス・クルシュカ症候群を疑うべきである.
臨床所見
- 前頭縫合隆起または頭蓋縫合早期癒合症
- 眼瞼下垂
- 非特異的形態変化(臨床像,頭蓋顔面症状を参照)
- 発達遅滞や自閉症状
頭部MRI所見.脳梁異常
分子遺伝学的検査
疑われる症状があり下記の分子遺伝学的検査所見のひとつが同定された発端者はワイス・クルシュカ症候群と確定診断される[Weissら 2017] (表1参照):
ZNF462遺伝子内のヘテロ接合性病的バリアント
ZNF462遺伝子を含む9q31.2領域のヘテロ接合性欠失
注: まれにZNF462遺伝子を切断する染色体転座の報告がなされている[Ramockiら 2003, Talisettiら 2003, Cosemansら 2018].
分子遺伝学的検査手法には表現型に応じて,遺伝子標的検査(単一遺伝子検査ならびに多遺伝子パネル検査)と包括的ゲノム検査(染色体マイクロアレイ検査,エクソームシーケンス検査,エクソームアレイ検査,ゲノムシーケンス検査)との組み合わせが含まれ得る.
遺伝子標的検査では臨床医がどの遺伝子が関与しているかを決定して行う必要があるが,ゲノム検査ではその必要はない.ワイス・クルシュカ症候群の表現型は広汎にわたるため,「疑われる所見」に記載されている特徴的所見を有する患者は遺伝子標的検査により診断される可能性が高い(選択肢1参照),一方で他の知的障害や非特異的形態変化を来す他の多くの遺伝性疾患と鑑別不能な表現型を有する患者はゲノム検査により診断される可能性が高い(選択肢2参照)
選択肢1
表現型所見からワイス・クルシュカ症候群の診断が示唆される場合,分子遺伝学的検査手法としては単一遺伝子検査または多遺伝子パネルの使用が考えられる.
- 単一遺伝子検査.ZNF462 遺伝子の配列解析により小さな遺伝子内欠失/挿入,ミスセンス,ナンセンスならびにスプライスサイトのバリアントが検出される; 一般的にはエクソンや全遺伝子の欠失や重複は検出できない.最初に配列解析を行う.病的バリアントが見られなかった場合には,遺伝子内の欠失や重複を検出するために遺伝子標的欠失・重複解析を行う.
- 染色体マイクロアレイ解析(CMA)では,配列解析では検出できないゲノムワイドな大きな欠失や重複(ZNF462 遺伝子を含む)を検出するため,オリゴヌクレオチドまたはSNPアレイを用いる.
- 知的障害多遺伝子パネルは,ZNF462 遺伝子と他の関連する遺伝子(鑑別診断を参照)を含み,CMAでは診断のつかない患者の遺伝学的要因を同定する可能性が最も高いが,基本的な表現型を説明することの出来ない遺伝子の臨床的意義不明のバリアントと病的バリアントとの鑑別には限界がある.注: (1)パネルに含まれる遺伝子と各遺伝子に用いられる検査診断精度は検査施設により異なり,時間経過より変化する可能性がある.(2)いくつかの多遺伝子パネルではこのGeneReviewsには記載されている症状とは関連のない遺伝子が含まれている可能性がある.ワイス・クルシュカ症候群の希少性を鑑みると,知的障害パネルの一部にはこの遺伝子が含まれていない可能性がある.(3)いくつかの検査施設ではパネル検査オプションには臨床医により特定された遺伝子を含む特注の検査室設計のパネルや特注の表現型対象のエクソーム解析が含まれる可能性がある.(4)パネル検査に用いられる手技には配列解析,欠失・重複解析のほか,配列解析以外の検査が含まれる可能性がある.本疾患には欠失・重複解析を含む多遺伝子パネル検査が推奨される(表1参照).
選択肢2
表現型が知的障害や非特異的形態変化が特徴の他の多くの遺伝性疾患との区別できない場合,包括的遺伝学的検査(どの遺伝子が関与している可能性が高いかを臨床医が判断する必要がない)が最良の選択肢である.エクソームシーケンスが最も一般的に用いられる; ゲノムシーケンスも可能である.
エクソームシーケンスで診断できない場合,そして特に常染色体顕性(優性)遺伝形式の根拠のある場合は,エクソームアレイ(臨床的に利用可能である場合)が配列解析では検出できない(複数の)エクソン欠失や重複を検出できると考えられる.
核型.表現型がワイス・クルシュカ症候群に合致するものの上記の検査ではZNF462 遺伝子を含む病的バリアントが検出されなかった場合には,標準的細胞遺伝学的解析によるZNF462 遺伝子を含む他の大きな細胞遺伝学的異常や染色体転座がないかの除外診断も考慮される[Ramockiら2003, Talisettiら 2003, Cosemansら 2018].
表1.ワイス・クルシュカ症候群に用いられる分子遺伝学的診断染色体座位と
| 遺伝子 1 | 検査法 | 検査により検出される病的バリアントを有する発端者の頻度2 |
|---|---|---|
| ZNF462 | 遺伝子配列解析 3 | 17/21 4 |
| 遺伝子標的欠失・重複解析5 | 不明 6 | |
| マイクロアレイ検査 7 | 2/8 8 | |
| 核型解析 | 2/8 9 |
- この遺伝子のアレルバリアントについては,分子遺伝学を参照.
- 遺伝子配列解析では良性,おそらく良性,意義不明,おそらく病的,病的なバリアントを検出する.バリアントには小さな遺伝子内欠失/挿入,ミスセンス,ナンセンスならびにスプライスサイトのバリアントが検出される; 一般的にはエクソンや全遺伝子の欠失や重複は検出できない.
- Kruszkaら [2019]
- 遺伝子標的欠失/重複解析では遺伝子内欠失や重複を検出する.実施される検査法の範囲には定量PCR,ロングレンジPCR,MLPA(multiplex ligation-dependent probe amplification)法,単一エクソンの欠失や重複を検出するよう設計された遺伝子標的マイクロアレイが含まれる.遺伝子標的欠失/重複検査では単一エクソンから全遺伝子にわたる欠失を検出しうる.しかしこれらの方法では,大きな欠失の切断点や隣接遺伝子の欠失は検出し得ない.
- 遺伝子標的欠失/重複解析の検出率のデータはない.
- 染色体マイクロアレイ解析(CMA)ではオリゴヌクレオチドやSNPアレイを用いて遺伝子配列解析では検出できないゲノムワイドな大きな欠失/重複(ZNF462遺伝子を含む)を検出する.欠失/重複の大きさの判定能力は,用いるマイクロアレイの種類や,9q31.2領域のプローブ密度に依存する.現在,臨床使用されているCMAは9q31.2領域を標的としている.
- Weissら[2017]
- Ramockiら [2003], Talisettiら [2003], Cosemansら [2018]
遺伝子レベルでの関連疾患
このGeneReviews内で述べられている以外の表現型でZNF462遺伝子の生殖細胞系列の病的バリアントに関連するものは知られていない.
臨床像
現在まで,21家系24名にZNF462遺伝子の病的バリアントが同定されてきた[Ramockiら 2003, Talisettiら 2003, Weissら 2017, Cosemansら 2018, Kruszkaら 2019].下記のこの疾患に関連する表現型症状の記載はこれらの報告例に基づく.
注: Ramockiら [2003] と Talisettiら [2003] は同一患者について報告している; 著者らはZNF462遺伝子を切断する均衡型転座により生じた融合タンパクが,この患者の症状を引き起こしていると推測していた.
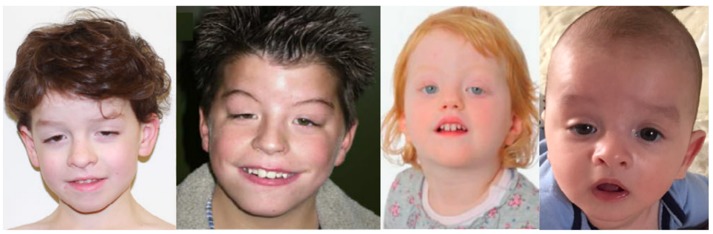
頭部顔面症状.最も一般的な顔面症状(図1):
- 眼瞼下垂 (20/24; 83%)
- 瞼裂斜下 (13/24; 54%)
- 目立つCupid’s Bow [上口唇上部縁の形状がキューピットの弓に似ることからCupid’s Bow と呼ばれる] (13/24; 54%)
- 弓状の眉 (12/24; 50%)
- 内眼角贅皮 (11/24; 46%)
- 球状の鼻尖を伴う短く上向きの鼻 (11/24; 46%)
半数以下の患者は前頭縫合隆起か前頭縫合かラムダ縫合の頭蓋縫合早期癒合を有する(9/24; 38%).
発達遅滞.大多数(既知の患者の75%以上)は全般性発達遅滞,運動発達遅滞,言語発達遅滞やこれらの組み合わせの何らかのタイプの発達遅滞を有する.
- 言語発達遅滞は最も多いな所見であり42%にみられる.
- 運動発達遅滞は次に多い所見であり38%にみられる.
- 筋緊張低下は運動発達遅滞の要因であり,50%の患者は筋緊張の低下を有する.
1/3(既知の24名の患者中8名)は自閉スペクトラム症を有する.
耳/聴力. 発端者の45%は聴力障害や外耳形態の異常を有する:
- 24名の患者中 6名(25%)が耳介低位
- 患者の 12/24 に,水平な耳輪脚,聳立耳,耳介陥凹,カップ耳,過剰に折れ込んだ耳などの耳介形態異常
- 24名の患者中に3名の様々な重症度の難聴
胃腸. 摂食問題は一般的で,全患者中の半数に摂食困難が報告さえており何名かは胃瘻チューブの設置を要した.摂食問題の内容は下記の通り.
- 胃食道逆流は Nissen噴門形成術を要する
- 経口摂取試行中の呼吸障害を来す喉頭軟化症
- 嚥下障害
- 好酸球性食道炎
- 咀嚼障害
心形態異常. 罹患者の少数(5/24; 21%)は,心室中隔欠損,大動脈二尖弁,大血管転位,動脈管開存などの先天性心形態異常を有する.
常. およそ25%の患者に軽度の四肢形態異常を認める.
- 3名は単一手掌横断襞を有する(13%).
- 3名は第V指屈指を有する (13%)
- 1名は拇指の近位付着を認めたと報告されているが,この患者は均衡型転座によりZNF462遺伝子とKLF12遺伝子の融合を有している[Cosemansら 2018].
脳梁形成不全は当初ZNF462遺伝子の機能喪失による主要症状と考えられていた; しかしながら多くの症例が確認されるにつれ,脳梁形成不全の患者の割合は25% (6/24)に近づきつつある.痙攣発作は現在までのところいずれの患者にも認められていない.
自然経過
ワイス・クルシュカ症候群の寿命が短縮するか否かは不明である.1名の報告症例は67歳である[Weissら 2017]. 障害のある多くの成人は先進的遺伝学的検査を受けていないため,この疾患の成人は十分に認知されておらず,十分に報告されていない可能性がある.
遺伝型と臨床型の関連
遺伝型・表現型連関は知られていない.
頻度
この疾患は希少であり,有病率は知られていない.21家系の24名の患者のみが知られている[Ramockiら2003, Talisettiら 2003, Weissら 2017, Cosemansら 2018, Kruszkaら 2019].
鑑別診断
表2.ワイス・クルシュカ症候群の鑑別診断として考慮すべき知的障害を伴う疾患
| 鑑別診断疾患 | 遺伝子 | 遺伝 形式 |
鑑別診断疾患の臨床症状 | |
|---|---|---|---|---|
| ワイス・クルシュカ症候群との重複症状 | ワイス・クルシュカ症候群との鑑別症状 | |||
| 眼瞼裂狭小,眼瞼下垂,逆内眼角贅皮 | FOXL2 | AD | 眼瞼下垂 耳介異常 弓状眉 |
瞼裂縮小 逆内眼角贅皮 小眼球症 斜視 |
| ヌーナン症候群 (Noonan syndrome) |
BRAF KRAS LZTR1 1 MAP2K1 NRAS PTPN11 RIT1 SOS1 |
AD (AR) 1 | 眼瞼下垂 耳介低位 先天性心疾患 |
眼間開離 発達遅滞はワイス・クルシュカ症候群より稀れ 低身長 翼状頸 |
| 遺伝性先天性眼瞼下垂 1 (OMIM 178300) | 未知 | AD | 眼瞼下垂 | 脳,頭蓋顔面,心形態異常はない |
| 遺伝性先天性眼瞼下垂2 (OMIM 300245) | 未知 | XL | 眼瞼下垂 | X連鎖遺伝形式 脳,頭蓋顔面,心形態異常はない |
| 三角頭蓋 I (OMIM 190440) | FGFR1 | AD | 三角頭蓋 | 軽度眉毛叢生 |
| 三角頭蓋 2 (OMIM614485) | FREM1 | AD | 三角頭蓋 | 一部の患者は小頭症 |
AD = 常染色体顕性(優性)遺伝; AR = 常染色体潛性(劣性)遺伝; XL = X連鎖遺伝
1. 常染色体潛性(劣性)遺伝の LZTR1遺伝子関連ヌーナン症候群が報告されている[Johnstonら 2018].
臨床的マネジメント
最初の診断時における評価
ワイス・クルシュカ症候群の診断を受けた患者の疾患の程度とニーズを確認するため,表3に総括した評価(診断のための評価の一部として行われなかった場合)が推奨される.
表3.ワイス・クルシュカ症候群の患者の初期診断後に推奨される評価| 器官/懸念事項 | 評価 | コメント |
|---|---|---|
| 頭蓋顔面 | 顔面形状と頭蓋縫合隆起を同定する理学的診断 | |
| 眼 | 眼科的評価 | 眼瞼下垂に対処 |
| 発達 | 発達評価 | 下記を含む:
|
| 精神医学/ 行動 | 精神神経学的評価 | 月齢12カ月以上: 自閉スペクトラム症を示唆する性格を含む行動の問題のスクリーニング. |
| 耳/聴覚 | 聴覚評価 | 難聴の評価 |
| 胃腸/摂食 | 胃腸科 / 栄養 / 摂食チーム評価 |
|
| 心血管 | 循環器科対診 | 基本的心エコー検査を推奨 |
| 神経学 | 神経学的評価 | |
| その他 | 臨床遺伝専門医や認定遺伝カウンセラー対診 | |
| 家族支援/社会資源 | 以下の必要性の評価:
|
表4.ワイス・クルシュカ症候群の患者の病変に対する治療
| 症状/懸念事項 | 対処 | 留意事項/その他 |
|---|---|---|
| 頭蓋縫合早期癒合症 | 頭蓋顔面チームや脳神経外科紹介 | 外科的修復の検討 |
| 眼瞼下垂 | 眼科医毎の標準的治療 | |
| 発達遅滞/知的障害 | 発達遅滞/知的障害の管理に関する項目を参照 | |
| 聴覚障害 | 補聴器が有用; 耳鼻咽喉科医毎の治療. | 早期介入や学区を通じての公的聴覚支援 |
| 摂食障害/嚥下障害/体重増加不良 | 摂食療法; 摂食問題が持続する場合には胃管留置も必要となることもある | 嚥下障害の臨床所見や症状がある場合には,臨床的摂食評価や放射線学的嚥下評価を早めに考慮する |
| 先天性心疾患 | 循環器科医毎の標準的治療 | |
| 家族/コミュニティ |
|
|
一次病変の予防
以下の情報は発達遅滞/知的障害を有する者へのアメリカ合衆国における一般的な推奨管理項目である; 標準的推奨事項は国により異なる場合がある.
発達遅滞/知的障害の管理事項
0〜3歳. 作業療法,理学療法,言語療法ならびに摂食療法利用のため,早期介入プログラムを紹介することが推奨される.アメリカ合衆国では早期介入は全州で利用できる連邦政府予算のプログラムである.
3〜5歳. アメリカ合衆国では地域の公立学区における発達支援就学前教育が推奨される.入園前に必要とされる援助や対処について評価が行われ,個別の教育計画(IEP)が策定される.
- アメリカ合衆国では,個人の能力レベルに基づきIEPを地域の公立学区が策定しなければならない.患児は21歳まで公立学区に留まることが許可される.
- 財政,就労/雇用,医療手配を含む移行計画に関する討議は12歳から開始すべきである.発達小児科医は成人期移行への支援を行うことができる.
全年齢. 適切なコミュニティ,州,教育機関の関与と両親が生活の質を最大限に保てるようにする支援を担保するために,発達小児科医と相談することが推奨される.
患者のニーズに基づいた個別の支援療法を検討することが推奨される.治療の方法に関する具体的推奨事項は発達小児科医からなされるであろう.
アメリカ合衆国では:
- 発達障害局(DDA)への登録が推奨される.DDAは 有資格者にサービスと支援を行う公的機関である.資格認定は州により異なるが,一般的には診断名や随伴する認知/適応障害により決定される.
- 収入や財源が限られている家族には補足的保証所得(SSI)が障害のある子どものために認定されることもある.
運動機能不全
粗大運動機能不全
- 可動性を最大化するために理学療法が推奨される.
- 必要に応じて耐久性のある医療器具の利用を考慮する(例えば,車椅子,歩行器,風呂椅子,装具,障害者用バギー)
微細運動機能不全. 摂食,保清,着替え,筆記などの適応機能に影響する微細運動機能の障害には作業療法が推奨される.
口腔運動機能不全. 経口摂取が安全と推定される場合,口腔運動制御不全のため摂食困難な患者には,一般的には作業療法士や言語療法士による摂食療法が推奨される.
コミュニケーションの問題. 言語表出に困難を有する者には代替コミュニケーション手段(例えば,補助代替コミュニケーション augmentative and alternative communication [AAC])の評価を考慮する.
社会/行動の問題
子どもたちは自閉スペクトラム症の治療に用いられている応用行動分析(applied behavior analysis (ABA))を含む介入を受け,恩恵を得られる可能性がある.ABA療法は個々の子どもの行動,社会性および適応面での長所や弱点を対象として行われるものであり,一般的には認定を受けた行動分析士により1対1で施行される.
経過観察
表5.ワイス・クルシュカ症候群の患者に推奨される経過観察
| 器官/懸念事項 | 評価 | 頻度 |
|---|---|---|
| 頭部 | 頭囲と頭蓋形状の評価 | 幼児期から小児期早期の評価時毎 |
| 眼 | 眼科的評価 | 眼瞼下垂の程度により頻度を決定 |
| 発達 | 発達状況と教育の必要性を観察 | 診察毎 |
| 耳 | 聴覚評価 | 臨床的に疑われる場合 |
| 摂食 | 身体成長指標の計測 栄養状態と経口摂取の安全性の評価 |
診察毎 |
| その他 | 社会福祉支援(例えば,レスパイトケアやその他の地元のサービス)と介護支援サービスに対する家族のニーズの評価 |
リスクのある親族の検査
遺伝カウンセリングを目的としたリスクのある近親者の検査に関する事項については,「遺伝カウンセリング」の項目を参照のこと.
研究中の治療法
アメリカ合衆国ではClinicalTrials.govに,ヨーロッパではEU Clinical Trials Registerにアクセスして,広汎な疾患に関する臨床研究の情報を検索すること.注: この疾患の臨床試験は行われていない可能性がある.
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ワイス・クルシュカ症候群は常染色体顕性(優性)遺伝形式で遺伝する.
家族構成員のリスク
発端者の両親
- ワイス・クルシュカ症候群と診断された患者の95%は新生のZNF462遺伝子の病的バリアントにより発症している.
- ワイス・クルシュカ症候群と診断された患者の5%には罹患した親がいる.家系内の臨床症状の多様性が報告されている; 罹患家系メンバーの症状は孤発性の眼瞼下垂から脳梁欠損および前頭縫合早期癒合にいたるまで表現型は多彩である[Weissら 2017, Kruszkaら 2019].
- 明らかな新生病的バリアントを有する発端者の両親には分子遺伝学的検査が推奨される.
- いずれの両親の白血球由来DNAにも発端者で検出された病的バリアントが検出されなかった場合,考えられる説明は発端者が新生病的バリアントであるか,一方の親の生殖細胞系列モザイク[性腺モザイク]である.ひとつの家系において親の生殖細胞系列モザイクが報告されている[Kruszkaら 2019].
- ワイス・クルシュカ症候群と診断された患者の何名かは,より軽症な家系員の疾患認知が行われずに,家族歴はなしとされるかも知れない.従って,発端者の両親の分子遺伝学的検査を行わずして明らかに家族歴なしとは確定できない.
発端者の同胞
発端者の同胞のリスクは,発端者の両親の遺伝学的状態に依存する.
- もし発端者の両親のいずれかがZNF462遺伝子の病的バリアントを有する場合,同胞にバリアントが伝わるリスクは50%である.家系内の臨床的多様性があるため,ZNF462遺伝子の病的バリアントが伝わった同胞の症状は発端者よりも重いかも知れないし,軽いかも知れない.
- もしZNF462遺伝子の病的バリアントが臨床的に非罹患者である両親いずれの白血球由来DNAにも認められなかった場合,親に生殖細胞系列モザイクが存在する可能性もあることから,同胞の再発確率は一般人口における確率よりわずかに高い[Kruszkaら 2019].
発端者の子
ワイス・クルシュカ症候群患者の子は各々,50%の確率でZNF462遺伝子の病的バリアントを受け継いで有する.
他の家族構成員
他の家系員のリスクは発端者の両親の状況に依存する; もし両親のいずれかがZNF462遺伝子の病的バリアントを有する場合は,その方の家系員にはリスクがある.
遺伝カウンセリングに関連した問題
明らかにde novo変異による家族 常染色体顕性(優性)遺伝疾患の発端者の両親のいずれにも病的バリアントが同定されていないか,疾患の臨床所見がない場合,病的バリアントは新生である可能性が高い.しかしながら,代理父や代理母(例えば,生殖補助医療によるもの)や非公開の養子縁組などの非医学的な要因も検討することができる.
家族計画
- 遺伝的リスクの確定や出生前/着床前遺伝学的検査の実施に関する検討を行う最適な時期は妊娠前である.
- 軽度発症の若年成人に遺伝カウンセリング(子の潜在的リスクや生殖の選択肢についての討議を含む)を行うことは適切である
DNAバンキング 検査手法論ならびに遺伝子や発症病理,疾患に関する我々の理解は将来進歩する可能性が高いため,分子診断で確認されていない(即ち発症の病理メカニズムが知られていない)発端者からのDNAバンキングは考慮されるべきである.
出生前診断
罹患家系構成員にZNF462遺伝子の病的バリアントが一旦同定されれば,リスクの増大した妊娠における出生前検査や着床前遺伝学的検査が可能である.しかしながら,ワイス・クルシュカ症候群には家系内での臨床症状の多様性があるため,分子遺伝学的検査結果では臨床症状を予測することは出来ない.
出生前検査を行おうとする医療専門家や家族の間でも見解の相違が存在する可能性がある.多くのセンターは出生前検査の利用を個人的な決定と考えるが,この懸念事項の議論も有用であろう.
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A. ワイス・クルシュカ症候群: 遺伝子とデータベース
| 遺伝子 | 染色体座位 | タンパク | HGMD | ClinVar |
|---|---|---|---|---|
| ZNF462 | 9q31.2 | Zinc finger protein 462 | ZNF462 | ZNF462 |
データは以下の標準的参照データベースより引用した: 遺伝子 HGNC; 染色体座位 OMIM; タンパク UniProt.
表B. ワイス・クルシュカ症候群のOMIM登録項目
| 617371 | ZINC FINGER PROTEIN 462; ZNF462 |
| 618619 | WEISS-KRUSZKA SYNDROME; WSKA |
序文. ZNF462は23個のジンクフィンガー・ドメインを有するジンクフィンガー・ファミリーに属するタンパクであり,DNAに結合し機能すると思われる[Changら2007].ZNF462は細胞分裂における卵割期に必要であり[Laurentら 2009],多機能細胞のクロマチン構造を維持に関与している[Masséら2010].
発症機構.ワイス・クルシュカ症候群は機能喪失機序により発症すると推定されている.疾患関連表現型発現の機序は不明である.病的バリアントの大部分はエクソン3に位置する[Kruszkaら 2019].
ZNF462-特異的研究事項. ZNF462遺伝子は12のトランスクリプトを有する.RefSeqデータベースにはZNF462遺伝子の2つのアイソフォームが登録されている: zinc finger protein 462 isoform 1 (NM_021224.6) は最長のトランスクリプトであり; isoform 2 (NM_001347997.1) は5' コード領域の代替インフレーム・スプライス部位を利用する.
更新履歴:
-
Gene Reviews著者:Paul Kruszka, MD, MPH.
日本語訳者: 沼部博直 (東京都立北療育医療センター)
GeneReviews最終更新日: 2019.10.31. 日本語訳最終更新日: 2023.04.11.[in present]
![]()