トリーチャー・コリンズ症候群
(Treacher Collins Syndrome)
[同義語: 下顎顔面骨形成不全症, Treacher Collins-Franceschetti症候群]
Gene Reviews著者: Sara Huston Katsanis, MS and Ethylin Wang Jabs, MD
日本語訳者: 江田 肖(瀬戸病院 遺伝診療科),石川亜貴(札幌医科大学医学部遺伝医学)
Gene Reviews 最終更新日: 2012.8.30 語訳最終更新日: 2015.1.6
この翻訳には新しい翻訳があります。こちらをクリック。
要約
疾患の特徴
トリーチャー・コリンズ症候群(TCS)は頬骨と下顎骨の形成不全、外耳奇形、下眼瞼欠損(亀裂)、下睫毛欠損、毛髪位異常(耳介前方の毛髪が頬まで生える)を 特徴とする 。患者の約40%~50%は耳小骨異常(耳小骨の硬化、形成不全や欠損)および中耳腔の形成不全による伝音性難聴を有する。内耳構造は通常正常である。この他、 より頻度の低い奇形として、 口唇裂を伴うあるいは伴わない口蓋裂、片側あるいは両側の後鼻孔狭窄/閉鎖がある。
診断・検査
トリーチャー・コリンズ症候群の診断は臨床所見およびX線画像所見に基づく。TCOF1(78%~93%)、POLR1C とPOLR1D(8%) は本症の原因遺伝子として知られている。
臨床的マネジメント
対症療法:治療は患者個々のニーズに応じる。集学的医療チームによる頭蓋顔面の管理が望ましい。新生児期に気道管理のため、特定体位や気管切開が必要となるかもしれない。難聴の治療には、骨伝導音声増幅器の使用、言語療法や教育的介入がある。頭蓋顔面再建はたびたび必要とされる。口蓋裂修復術(必要があれば)は生後1年から2年の間に、頬骨と眼窩の再建は5歳~7歳の間に、外耳再建は6歳以降に行う。上下顎の再建時期は重症度によるが、下顎矯正は通常16歳以前に行う。両側性小耳症および/または外耳道狭窄は再建を 必要とす る。
研究中の治療法:頭蓋顔面形成異常部に幹細胞を移植することによって外科的治療効果の向上が期待されている。しかし、これらの術式はまだ実験段階であり、議論の余地が残っている。また、胚発生時のp53遺伝子発現を抑制することで、神経堤関連障害の予防になるかもしれない。
遺伝カウンセリング
本症のほとんどは常染色体優性遺伝であるが、一部(~1%)は常染色体劣性遺伝形式をとる。
常染色体優性TCS:患者の約60%は新規突然変異である。患者の子は50%の確率で同じ遺伝子変異を受け継ぐ。事前に家系内の病的変異が同定されていれば、出生前診断は可能である。
診断
臨床診断
本症の診断は臨床所見およびX線画像所見に基づく。
臨床所見の鑑別[Hertle et al 1993, Posnick & Ruiz 2000, Marszalek et al 2002, Teber et al 2004, Trainor et al 2009]
- TCOF1遺伝子変異を有する患者の写真はFigure 1を参照する。患者の詳細やほかの写真はFigure 2を参照する。
- 本症の臨床所見が明白でない8名の患者のうち3名はTCOF1遺伝子変異を認めなかった。Figure 3を参照する。
- 同じ家系の患者特徴はFigure 4を参照する。
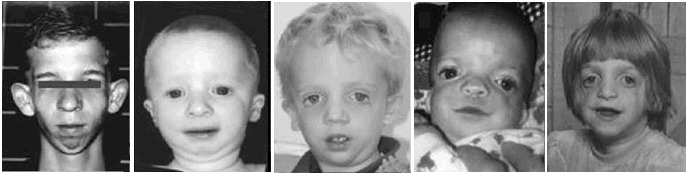
Figure 1. TCOF1遺伝子変異を認めた患者
Nature Publishing Group[Teber et al 2004]から複製許可を取得済み。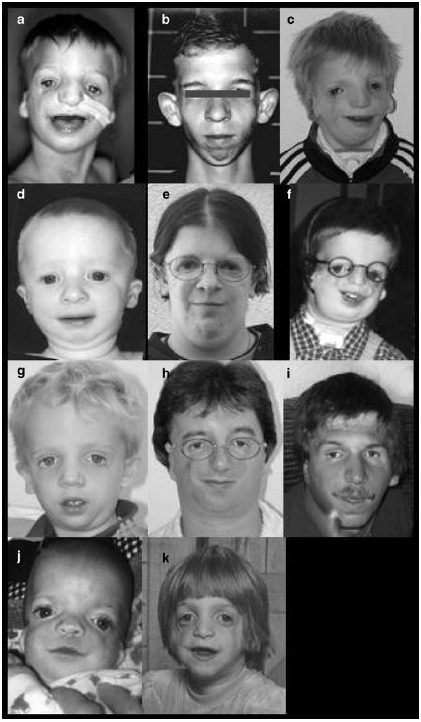
Figure 2. TCOF1遺伝子変異を認めた患者(a-k)。 写真はTCOF1遺伝子変異の場所の順に 並べている。
a. M17639患者:変異Met1Ile
b. M17807患者:121Ter
c. M20194患者:209Ter
d. M19371患者:Lys367Ter
e. M18982患者: Gln563Ter
f. M18923患者: 795Ter
g. M17629患者: Gln818Ter
h. M18013患者: 854Ter
i. M18774患者: Gly848Ter
j. M17995患者: c.2629-3A>G
k. M18293患者: 1392Ter
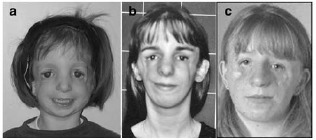
Figure 3. 本症の臨床的特徴が明白でない8名の患者のうち3名はTCOF1遺伝子変異を認めなかった。
a. M17739患者
b. M18662患者
c. M17652患者
Nature Publishing Group[Teber et al 2004]から複製許可を取得済み。
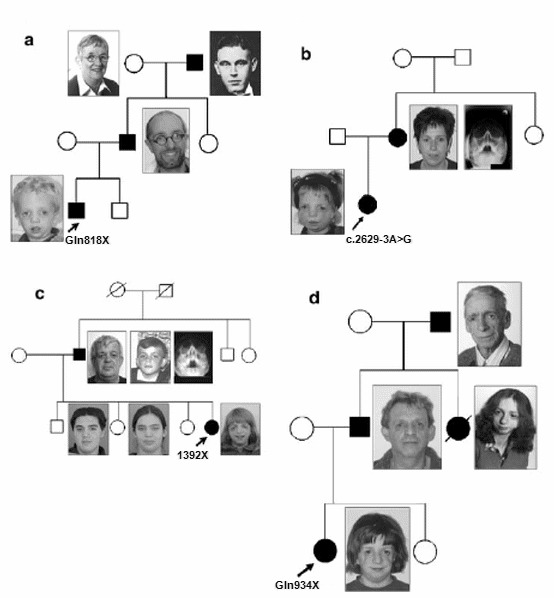
Figure 4. 同一家系内の患者
a. 患者M17629の家系図。発端者は眼瞼裂斜下、頬骨複合体の低形成、軽度な耳形成異常との特徴的顔貌、伝音性難聴と下顎の低形成を有する。この家系にはナンセンス変異(Gln818X)が同定されている。発端者の父親の臨床所見は軽く、顎鬚と眼鏡によって、表現型は更に不明瞭となっている。父親は伝音性難聴がないが、耳の形成異常は手術治療を受けた。父方祖母は息子に近い顔貌的特徴を持ち、耳の形成異常の家族歴もある。驚くことに、父方祖父は、遺伝子変異を持っているにもかかわらず、本症の特徴的顔貌を見られなかった。祖父は検査や画像診断を拒否したが、本症の浸透度が不完全である実例となるかもしれない。
b. 患者M17995の家系図。発端者は頬骨複合体低形成、両側性外耳道閉鎖を伴う小耳症、口蓋裂、両側性後鼻孔閉鎖を有する。スプライシングサイト変異(c.2629-3A>G)は原因と考えられている。患者の母親には下顎骨の低形成を認め、眼瞼裂の傾斜は正常であるが、臨床的に右頬骨複合体低形成を 疑われていた。X線画像診断(ウォータース法)では頬骨複合体低形成が確認された。この母親の表現型は軽度と考えている。
c. 患者M18293。発端者は両側性小耳症、頬骨複合体低形成および眼瞼裂斜下を有する。写真から発端者の父親は非罹患と判断したが、後の遺伝子検査で保因者であることが分かった。父親の顔貌的特徴は僅かに下方へ傾斜する眼瞼裂だけであった。軽度な顔貌的特徴の鑑別は小児期で行うのははるかに分かりやすい。ウォータース法では、父親の両側性頬骨複合体の低形成および左側の頬骨弓の無形成を認めた。発端者の兄弟は同じ遺伝子変異を受け継いでいなかった。d. M22186家系。発端者は重度な表現型を有する。発端者の父親には軽度な頬骨複合体低形成だけを認めた。父親は自分が罹患していないと確信していたが、罹患した娘によって父親の診断がついた。父親の姉は重度な表現型を有し、心不全のため20歳で死亡した。父方祖父には軽度な難聴と眼瞼裂斜下を認めた。遺伝子検査ができなかったが、この祖父は罹患者である可能性が高い。
大症状
- 頬骨と下顎骨形成不全 [Posnick 1997]は下記の症状を引き起こす。
- 両側性凸状の顔面輪郭を伴う顔面中央形成不全(89%)、高い鼻骨および特徴的な眼窩外側の形成不全による下方に傾斜する眼。
- 小顎症と下顎後退症(78%)、およびこれらによる顎関節と顎筋の様々な症状。
- 外耳形成異常(77%) 外耳の欠損、低形成または形成異常(小耳症)、捻転耳を含む。
- 下眼瞼形成異常は下記を含む。
- 欠損(亀裂)(69%)
- 疎な睫毛、あるいは睫毛の部分欠損や完全欠損(53%)
- 常染色体優性遺伝形式の家族歴(40%)
小症状
- 外耳道狭窄あるいは閉鎖を含む外耳形成異常(36%)
- 伝音性難聴(40%-50%) ほとんどの場合は耳小骨の硬化、形成不全や欠損、または中耳腔の形成不全が原因である。内耳構造は通常正常である。
- 眼科的異常
- 視力喪失(37%)
- 弱視(33%)
- 屈折異常(58%)
- 屈折不同(17%)
- 斜視(37%)
- 口唇裂を伴うあるいは伴わない口蓋裂(28%)
- 耳介前毛髪変位(26%) 耳介の前方から頬骨の外側まで毛髪が生える。
- 気道異常は下記を含む
- 気道婁孔
- 片側あるいは両側の後鼻孔狭窄/閉鎖
- 運動や言語発達遅滞
- 常染色体劣性遺伝形式の家族歴(1%)
X線所見の鑑別
- 頬骨弓の低形成あるいは無形成は後頭オトガイのX線検査で確定される。下顎骨形成不全やほかの異常を確認するために、X線検査には頭蓋骨の後頭オトガイの投影図(ウォーターズ法)と正写図を含む。
- 頬骨形成不全はCTによる眼窩内測量で平均値を得ても、頬骨測量で正常値より低い [Posnick & Ruiz 2000]。
- 顔面弓隆による下顎骨後退症の程度は、頭蓋X線撮影で確認する。
分子遺伝学的検査
遺伝子
トリーチャー・コリンズ症候群の原因遺伝子は、TCOF1,POLR1C,POLR1Dの三つとして知られている(Table 1を参照する)。
その他の遺伝子座不均一性の可能性
本症の典型的臨床所見を有する一部の患者にはTCOF1,POLR1C,POLR1Dいずれの遺伝子変異も認めなかった。
臨床検査
Table 1. トリーチャー・コリンズ症候群の分子遺伝学的検査
| 遺伝子1 | 当該遺伝子変異によるトリーチャー・コリンズ症候群の割合 | 検査方法 | 検出される変異2 |
|---|---|---|---|
| TCOF1 | 71%-93%3 | シークエンス解析4 | 遺伝子配列変異 |
| 欠失/重複解析5 | (複数の)エクソンあるいは全遺伝子欠失6 | ||
| POLR1D POLR1C |
本症患者の8%はTCOF1遺伝子変異を検出されなかった7 | 欠失/重複解析5 | (複数の)エクソンあるいは全遺伝子欠失7 |
| シークエンス解析4 | 遺伝子配列変異 | ||
| シークエンス解析4 | 遺伝子配列変異 | ||
| 欠失/重複解析5 | (複数の)エクソンあるいは全遺伝子欠失8 |
- 遺伝子座位またはタンパク質はTable A.遺伝子とデータベースを参照する。
- アレル変異は分子遺伝学の項を参照する。
- 本症患者の大多数はヘテロ接合のTCOF1遺伝子変異が原因である。 [Treacher Collins Syndrome Collaborative Group 1996]。Splendoreら [2000]は93%、Teber[2004]らは78%の精度でTCOF1遺伝子に変異がないが明白なトリーチャー・コリンズ症候群所見の患者を報告した。Bowmanら[2012]はトリーチャー・コリンズ症候群と強く疑われている血縁関係のない119名の患者のうち、84名がTCOF1遺伝子病的変異 を同定した(70.6%)。
- シークエンス解析で検出できる変異は微小な遺伝子内欠失/挿入、ミスセンス、ナンセンス変異、スプライスサイト変異がある。一般的に、エクソンや全遺伝子欠失/重複が検出できない。シーケンス解析結果の解釈はhere.を参照する。
- 欠失/重複解析とは、シークエンス解析ではなかなか検出できない、ゲノムDNAの翻訳領域やイントロン隣接領域における遺伝子変異を同定するための検査であり、変異が起こった遺伝子/染色体断片を含む、定量的PCR、ロングレンジPCR、MLPA、または染色体マイクロアレイ(CMA)など様々な方法が含まれる。
- Beygo et al [2012], Bowman et al [2012]
- Dauwerse et al [2011]
- POLR1C遺伝子の欠失や重複によるトリーチャー・コリンズ症候群の報告はない。(注:定義上、欠失/重複解析はゲノムDNAシーケンス解析で検出できない染色体再構成を検出する)
検査手順
発端者の確定診断のための検査 少なくとも大症状の2つ、あるいは小症状の3つを有する患者には、分子遺伝学的検査を考慮すべきである。
- 下記の患者には、最初にTCOF1遺伝子のシーケンス解析および欠失/重複解析を行う。
- 本症の常染色体優性遺伝形式の家族歴、
および
- 孤発例(例:家系内罹患者は1人しかいない)
- TCOF1遺伝子に変異や欠失を認めなければ、次にPOLR1D遺伝子シーケンス解析を考慮する。
- 下記を当てはまる場合、POLR1C遺伝子シーケンス解析を考慮する。
- 複数の同胞が罹患し、および/あるいは近親婚の家系、
または
- TCOF1とPOLR1D遺伝子変異を認めなかった孤発例
出生前診断および着床前診断(PGD)は事前に家系内における病的変異が同定される必要がある。
遺伝学的関連(アレル)疾患
TCOF1,POLR1D,POLR1Cにおける遺伝子変異はほかの表現型に関与していない。
臨床像
自然経過
トリーチャー・コリンズ症候群(TCS)は家系間および家系内に明らかな臨床的多様性が見られる[Posnick & Ruiz 2000, Teber et al 2004]。表現型が軽度で診断がついていない患者もいれば、重度な顔面形成異常や生命に関わるような気道障害をもつ患者もいる [Edwards et al 1996]。
本症の典型的所見は出生時に見られ、両側性且つ対照性 である。
トリーチャー・コリンズ症候群の新生児は、気道狭窄あるいは極端に短い下顎骨による重症な小顎症の場合、気道管理が必要となる。新生児期の気道閉鎖は後鼻孔の閉鎖、狭窄あるいは舌沈下を伴う重症な小顎症による場合がある。新生児死亡の多くはこれらの奇形による閉塞性睡眠時無呼吸が原因である。
本症に見られる伝音性難聴の原因は耳小骨低形成や中耳腔欠損の中耳異常である。内耳構造は通常正常である。患者の外耳異常は通常外耳欠損、小耳症や捻転耳であるが、一部の患者には外耳道の閉鎖や狭窄も見られる。
下眼瞼欠損を含む眼科的異常は患者の大多数に見られ、角膜を守るための措置を行うべきである。屈折異常、屈折左右不同、斜視を伴う視力喪失の可能性がある。
頭蓋縫合早期癒合症は本症の症状ではないが、頭蓋は異常な形(両側狭窄の短頭症型頭蓋)を呈する可能性がある[Posnick 1997]。
Da Silva Dalben et al [2006] は患者の60%が歯牙異常を有し、1人の患者には1個から8個の異常があると報告した。確認された異常は歯発育不全(33.3%)、エナメル斑(20%)、上顎第1大臼歯の異所芽出(13.3%)を含む。
より低い頻度で見られる所見は下記となっている。
- 鼻骨の変形
- 高いアーチ状の口蓋
- Angle分類クラスIIの前方開咬(不正咬合)
- 視力喪失(37%)
たまに見られる所見は下記となっている。
- 上眼瞼の欠損[Marszalek et al 2002]
- 眼間開 離 [Marszalek et al 2002]
- 後鼻孔閉鎖
- 巨口症
外耳道異常の有無および重症度は、中耳異常の有無と重症度との強い相関を認める [Posnick 1997]。
軽度な発達遅延との報告はあるが、知能は通常正常である。
生殖能力は正常である。
遺伝子型‐表現型の相関
遺伝子型から表現型を予測するのは不可能である [Edwards et al 1997, Splendore et al 2000, Teber et al 2004, Schlump et al 2012]。
Teberら[2004]のデータによると、TCOF1の3'翻訳領域に変異がある患者の伝音性難聴の頻度は低い。同じ遺伝子変異 (c.790_791delAG, p.Ser264GlnfsTer7) を有する4名の患者の表現型の比較によれば、TCOF1遺伝子変異の患者に見られる遺伝子発現の違いは、遺伝的、環境的および統計学的素因の組み合わせに影響される[Schlump 2012]。
浸透率
本症の浸透率 は高いが、遺伝子変異を有するにも関わらず発症しない症例も報告されている。
- Marresら[1995]のマッピング研究によって、 不完全浸透が分かった(例、TCOF1遺伝子変異を持つ患者は本症の臨床所見あるいはX線画像所見がない)。臨床所見および連鎖解析に基づき、Marresらは初めて遺伝子変異を有するが、表現型のない1人の患者を確認された。
- Katsanisら[2003]は罹患者である母親と息子に同じTCOF1 c.4369_4373delAAGAA の遺伝子変異を確認した。その後の妊娠では出生前検査が実施され、胎児に同じ遺伝子変異を認めたが、母親と第一子に見られた眼瞼裂斜下と下眼瞼欠損、下顎骨低形成、小耳症のような臨床所見のない第二子を出産した。この症例から、稀ではあるが、特に出生前診断においては、表現型の多様性および浸透率 の不完全を考慮すべきである。
- TCOF1遺伝子変異 c.2490delA (exon 15) と c.2853dupT (exon 16)を認めた罹患児の両親に浸透率 の不完全はDixonら[2004]に報告されている
- 発端者にPOLR1D遺伝子変異が確認された4家系において、この変異はほかの家系員に浸透率 の不完全が見られた [Dauwerse et al 2011]。
表現促進
世代を経て表現型が明らかに重症化する現象は診断バイアスに起因している。 Splendore et al [2002] は発端者が両親より耳形成異常の頻度が高いため、耳の異常は医学的評価に重要な指標の1つであると述べている。
用語
常染色体優性トリーチャー・コリンズ症候群はFransceschetti-Zwahlen-Klein 症候群、または頬骨下顎骨異形成とも呼ばれている。
頻度
本症の頻度は1万人から5万人に1人と推測されている [Fazen et al 1967, Argenta & Iacobucci 1989, Gorlin et al 2001, Trainor et al 2009]。
鑑別診断
下顎骨顔面形成不全症はほかに、Toriello症候群 (OMIM 301950), Bauru症候群 (OMIM604830), Hedera-Toriello-Petty症候群 (OMIM 608257), 及び Guion-Almeida症候群がある [Wieczorek et al 2009]。
トリーチャー・コリンズ症候群の特徴はNager症候群 (OMIM 154400)、Miller症候群 (OMIM 263750) 、Goldenhar症候群 (OMIM 164210)、Pierre Robinシーケンス(OMIM261800)と類似する。
Nager症候群とMiller症候群の場合、患者は下顎骨形成不全のほかに、四肢の奇形も見られる。
トリーチャー・コリンズ症候群の眼瞼欠損は下眼瞼に見られ、通常対照的であるが、Goldenhar症候群の眼瞼欠損は上眼瞼に見られ、対照的でない場合がある。
トリーチャー・コリンズ症候群と異なり、Pierre Robinシーケンスの関連所見(小顎症、舌沈下、口蓋裂を伴うあるいは伴わない気道障害を含む)は治療介入がなくても自己治癒する傾向がある [Singh & Bartlett 2005]。
非症候群性の下顎低形成患者には重度な下顎障害(顎関節硬直、無舌/小舌、稀に頭蓋顔面裂を含む)および進行性小顎症あるいは下顎後退が見られる。研究によれば、先天性下顎骨低形成を有する266人のうち52人はトリーチャー・コリンズ症候群であった[Singh & Bartlett 2005]。しかし、これらの患者には分子学的診断を行っていない。
注:本症に関する鑑別診断は、診断支援ソフトを参照すること(事前登録または研究目的の使用が必要)。
臨床的マネジメント
初診時の評価
トリーチャー・コリンズ症候群と診断された乳児に、下記の評価項目は推奨されている。
- 後鼻孔閉鎖/狭窄および/あるいは小顎と舌沈下による中咽頭障害
- 口蓋裂
- 嚥下機能
- 正式な聴力検査による聴力評価(難聴および遺伝性難聴概説を参照する)
- 眼球外運動、角膜上皮および視力に注意した眼科的評価
出生後6ヶ月以内の難聴なら、頭部、頸部および外耳、中耳、内耳の解剖学的情報を得るために頭蓋顔面CTスキャンを実施する。
歯牙異常に対する評価は歯が芽出後に行う。
治療法
患者個々のニーズに応じる治療を行う。臨床遺伝医、形成外科医、頭頸部専門医、耳鼻咽喉科医、歯科医、言語聴学士、心理士による集学的管理チームが望ましい。
主な管理項目は三つの年齢および重症度によって分かれる [Hayashi et al 2007,Thompson et al 2009]。
- 生後2歳まで:気道障害と哺乳障害
- 3歳~12歳:言語療法を取り組まれた教育システム
- 13歳~18歳:下顎矯正術
気道に対する外科的介入は標準的で、主に呼吸機能の改善や鼻孔の再建、下顎骨の伸長となる [Kobus & Wojcicki 2006, Jayasekera 2007]。妊娠中に本症と診断された場合、出生時の気道管理を考慮する [Morillas et al 2007]。新生児期の気道管理は通常、乳児の特定体位や気道瘻孔造設を含む。適切な管理を行っていれば、患者の寿命は一般集団と変わりがない。
気道保護しながら、適切なカロリー摂取を確保するために、胃瘻造設が必要となるかもしれない [Marszalek et al 2002]。
難聴の治療には、骨導拡大、言語療法および教育的介入がある。埋め込み型骨導補聴器(bone-anchored hearing aid, BAHA)は1つの選択肢である[Marres 2002]。
頭蓋顔面再建はたびたび必要となる [Posnick 1997, Zhang et al 2009]。一般に、骨再建には、 軟部組織の 修正を先に行う。 再建は顔面不対称 の進行を予防する。Posnick[1997]は再建術に関して、下記を推奨している。
- 口蓋裂の修復は1~2歳に行う [Kobus & Wojcicki 2006]
- 頬骨と眼窩の再建は頭蓋眼窩頬骨の骨発生が完成した後に行う(5~7歳まで)
- 上下顎の再建
- Type I(軽度)とType IIA(中度)形成異常:13~16歳
- Type IIB(中度~重度な形成異常):骨成長成熟時
- Type III(重度な形成異常):6~10歳
下顎矯正術は通常16歳までに行う。
歯並びはたびたび矯正が必要となる。
外耳再建は6歳に過ぎてから行うべきである。また、外耳道や中耳の再建を優先して行うべきである。
外耳道と中耳の再建は両側の小耳症および/または耳道狭窄の患者に行うべきである。
必要があれば、下眼瞼欠損はボツリヌス毒素とその後の 手術で対応する[Warner et al 2008]。
リスクのある血縁者の評価
遺伝カウンセリング目的の血縁者に関する検査は「遺伝カウンセリング」の項目を参照する。
研究中の治療
頭蓋顔面異常、特に、骨と軟骨に対する外科治療の効果は幹細胞の移植によって改善されるかもしれない。しかし、これらの術式はまだ実験段階で、議論の余地が残っている[Trainor et al 2009]。
胚形成時のp53遺伝子発現抑制は神経堤障害を予防する効果があるかもしれないと言われているが [Jones et al 2008, Trainor et al 2009]、同じく議論の余地が残っている。
より広範の疾患の臨床研究に関する情報はClinicalTrials.govを参照すること。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
トリーチャー・コリンズ症候群(TCS)はTCOF1遺伝子(またはより頻度の低いPOLR1D遺伝子)のヘテロ接合変異が原因の常染色体優性遺伝性疾患である。本症のほとんどは常染色体優性遺伝形式をとる。
POLR1C遺伝子における複合ヘテロ接合変異が原因のTCSは常染色体劣性形式で受け継がれる。
患者家族のリスク
発端者の両親―常染色体優性遺伝
- 常染色体優性TCS患者の約40%は罹患者の親がいる。
- 常染色体優性TCS発端者の約60%は新規突然変異である [Jones et al 1975, Splendore et al 2000, Trainor et al 2009]。
- 明らかな新規突然変異である発端者の場合、その両親に対する評価は、発端者で認められた変異に対する分子遺伝学的検査、発端者に遺伝子変異を認めなかった場合、X線検査(ウォータース像)は軽度な頬骨弓形成不全や無形成を検出できるかもしれない [Marres 2002]。
注:常染色体優性TCS患者の約40%は罹患者の親がいるものの、表現型が軽度で、稀に浸透率 が不完全のため、家族歴が見られない場合がある。
発端者の同胞
- 発端者の同胞のリスクは発端者の両親の遺伝学的状況 による。
- 発端者に罹患者の親がいれば、発端者同胞のリスクは50%となる。しかし、表現型や重症度は予測できない。
- 両親のいずれも病的変異を認めなかった場合、親の生殖細胞系列モザイクか発端者の新規突然変異、二つの可能性が考えられる。
- TCOF1遺伝子1639_1640delAGにおける生殖細胞系列モザイクはShooら[2004]に報告されている。しかし、この変異はトリーチャー・コリンズの子を持つ非罹患の母親の皮膚繊維芽細胞ではなく末梢血で検出されている。
- 非罹患の両親から罹患児二人が生まれたとの報告はあるが、本症の遺伝的不均一性、生殖細胞系列モザイクあるいは検出できなかったTCOF1遺伝子によるものなのかは不明である[Splendore et al 2000]。
発端者の子
- 常染色体優性TCS患者の子は50%の確率で同じ変異を受け継ぐ。
- しかし、表現型やそれらの重症度は予測できない。
他の血縁者
家系内ほかの血縁者のリスクは発端者の両親の遺伝学的状況 による。発端者に罹患者あるいは本症の原因遺伝子に変異を有する親がいれば、その親の血縁者は同じ変異を有する可能性がある。
発端者の両親―常染色体劣性遺伝
発端者の両親
- 常染色体劣性遺伝TCSの子を持つ両親は必ずヘテロ接合となる(例:一本のPOLR1C変異アレルを持つ保因者)
- ヘテロ接合(保因者)は発症しない
発端者の同胞
- 理論的に、発端者の同胞は25%の確率で本症を発症し、50%の確率で発症しない保因者となり、25%の確率で変異アレルを受け継がない。
- リスクのある同胞は罹患していないことが分かっていれば、彼/彼女が保因者となる確率は2/3となる。
- ヘテロ接合(保因者)は発症しない。
発端者の子 常染色体劣性遺伝TCS患者の子は必ずPOLR1C病的変異のヘテロ接合(保因者)である。
ほかの血縁者 発端者の両親の同胞は50%の確率で保因者になる。
保因者検査
家系内におけるPOLR1C病的変異が同定されていれば、リスクのある血縁者の保因者検査は可能。
遺伝カウンセリングに関連した問題.
明らかな新規突然変異の家系
常染色優性遺伝TCS発端者の両親には病的変異や本症の臨床所見を認めなければ、発端者は新規突然変異である可能性が高い。 しかしながら,生物的父親や生物的母親が異なる場合(例:生殖補助医療による)あるいは,明かされていない養子縁組など,非医学的な要因が関係する可能性もある。
家族計画
- 意思決定に至るまで、妊娠の前に遺伝学的リスク、保因者や出生前診断の適応を話し合うための十分な時間をとる。
- 罹患者、保因者あるいは保因者になり得る若い成人に遺伝カウンセリング(子の潜在的リスクや生殖の選択肢に関する話し合いを含む)が行われることが望ましい。
DNAバンク
DNAバンクは主に白血球から調製したDNAを将来の使用のために保存しておくものである。検査法や遺伝子,変異あるいは疾患に対するわれわれの理解が進歩するため,罹患者のDNAを保存することは考慮すべきかもしれない。
出生前診断
分子遺伝学的検査 家系内における病的変異が分かっていれば、リスクのある妊娠に対する出生前診断は羊水検査(妊娠15-18週)または絨毛検査(妊娠10-12週)から採取した胎児DNAを用いて行うことができる。
注:(1)TCS病的変異に対する出生前診断は本症の表現型や重症度を予測することができない。(2)遺伝カウンセリングを提供時や出生前検査結果説明時に、TCOF1遺伝子の4369_4373delAAGAA変異の不完全な浸透度は必ず考慮する。
*妊娠週数は最終月経の開始日あるいは超音波検査による測定に基づいて計算される。
超音波検査 リスクのある妊娠に対し、超音波検査による羊水過多、小頭症、胎児顔面形成異常(前 額の傾斜、小眼球、小顎)や胎児嚥下運動異常の出生前診断は可能 [Rotten et al 2002, Tanaka et al 2002]。しかし臨床所見が軽度な場合、見逃される可能性がある。
本症のような知的に影響がなく、治療法のある疾患に対し、出生前検査は通常行わない。出生前検査を行うことに関しては、 特にその目的が早期診断でなく中絶を考慮している場合,医療従事者内でも家族内でもさまざまな意見があるだろう.出生前診断の決定を両親の選択として捉えている医療機関がほとんどであるが,これらの問題について慎重な議論を行うことが望ましい.
着床前診断(PGD) 家系内の遺伝子病的変異が同定されている場合には技術的には可能である。
訳注:日本では本疾患に対して出生前診断の適応があるとは考えられていないし,着床前診断も行われていない。
関連情報
ひまわりの会~トリーチャー・コリンズ&ネイガー症候群の親の会
www.doremi-hochouki.com/himawari
分子遺伝学
下記の記述は最新の情報が含まれているため、GeneReviewsに記載されているほかの情報と異なる場合がある
Table A トリーチャー・コリンズ症候群:遺伝子とデータベース
| 遺伝子記号 | 遺伝子座 | タンパク質 | 座位特異性 | HGMD |
|---|---|---|---|---|
| TCOF1 | 5q32 | Treacle protein | TCOF1遺伝子変異データベース 難聴遺伝子変異データベース(TCOF1) TCOF1 database |
TCOF1 |
| POLR1C | 6q21.1 | Obscurin-like protein 1 | POLR1C @LOVD | POLR1C |
| POLR1D | 13q12.2 | Coiled-coil domain-containing protein 8 | POLR1D @LOVD | POLR1D |
Table B OMIMにおけるトリーチャー・コリンズ症候群関連情報
| 154500 | TREACHER COLLINS SYNDROME 1; TCS1 |
| 248390 | TREACHER COLLINS SYNDROME 3; TCS3 |
| 606847 | TCOF1 GENE; TCOF1 |
| 610060 | POLYMERASE I, RNA, SUBUNIT C; POLR1C |
| 613715 | POLYMERASE I, RNA, SUBUNIT D; POLR1D |
| 613717 | TREACHER COLLINS SYNDROME 2; TCS2 |
TCOF1
遺伝子構造
TCOF1遺伝子には27個のエクソンが含まれている。この中にはフレーム内の選択的スプライシングを受ける3つのエクソン(6A,16Aと19A)と 、 3'非翻訳領域を含むエクソンが含まれている [So et al 2004]。最も長い転写物(NM_001135243.1) はエクソン1から始まり、4,467個のヌクレオチドの翻訳領域を含む。この転写物には93-bpの5'非翻訳領域と507bpの 3'非翻訳領域がある[Dixon et al 1997a]。より詳細な遺伝子およびタンパクの情報は、Table A、遺伝子記号を参照する。
正常アレル変異 明らかに病的でない遺伝子多型 (≥18) と稀な遺伝子変異 (≥17)は確認されている [Splendore et al 2000, Ellis et al 2002, Splendore et al 2002, Dixon et al 2004,Su et al 2007]。TCOF1遺伝子変異データベースを参照する[Splendore et al 2005]。 病的アレル変異 多くのTCS家系において、新しい変異は確認されている。その種類は数百以上にのぼる [Gladwin et al 1996, Treacher Collins Syndrome Collaborative Group 1996,Edwards et al 1997, Wise et al 1997, Splendore et al 2000, Ellis et al 2002, Splendore et al 2002, Dixon et al 2004, Horiuchi et al 2005, Trainor et al 2009, Bowman et al 2012]。現在同定されていた変異の大多数は挿入や欠失によるフレームシフト変異である。その結果として、転写が途中で中断し、正常より短い転写物が生成される。変異の場所は全遺伝子に点在する。TCOF1遺伝子変異の中に、57%は小規模な欠失や挿入、16%はスプライシングサイト変異、23%はナンセンス変異、4%はミスセンス変異である [Bowman et al 2012]。1個以上のエクソンが関与する大規模な欠失は患者の5%に見られる [Beygo et al 2012, Bowman et al 2012]。同義変異が エクソンのミススプライシングを引き起こした1症例が 報告されている [Macaya et al 2009]。しかし、既知の遺伝子変異を検出する 方法として 、ヌクレオチド置換は 過小評価される可能性がある。
1回以上に現れた変異はいくつかがあるが、c.4369_4373delAAGAAの再現性は最も高い変異である。この変異はTCOF1遺伝子変異を認めた患者の16%を占める。
Table 2 本章に記載されているTCOF1病的アレル変異
| DNAヌクレオチドの変化 (Alias¹) |
アミノ酸の変化 (Alias1) |
基準配列 |
|---|---|---|
| c.790_791delAG | p.Ser264GlnfsTer7 | NM_001135243?.1 NP_001128715?.1 |
| c.2490delA | p.Val831Ter | |
| c.2853dupT (2853_2854insT) |
p.Ala952CysfsTer5 | |
| c.4369_4373delAAGAA | p.Lys1457GlufsTer12 |
注:上記に記されている変異は著者が提示したものであり、GeneReviewsはその内容に対し、検証を行っていない。
- この変異名は最近の命名法に従わない。
正常遺伝子産物
144kdのトリークル(Treacle)タンパクは1488個のアミノ酸からなる。Treacleは低複雑性、ユニークなN末端とC末端を持つ、3ドメインの核小体タンパクである。このタンパクは構造的に核小体リン酸化タンパクNopp140に関与する [Isaac et al 2000]。中心にある 10個の繰り返し モチーフはプロティンキナーゼCとカゼインキナーゼ2リン酸化サイトを含む[Dixon et al 1997b, Winokur & Shiang 1998]。このタンパクはC末端に少なくと も2つの機能的核輸送 シグナルおよび1つの核小体輸送 シグナルを有する 。Nopp140とTreacleはLIS1モチーフを含んでいることから、微小管動態への関与が 推測される[Emes & Ponting 2001]。核小体低分子リボ核タンパク質hNop56pと相互作用することから、Treacleはリボソーム生合成への関与が示唆される[Hayano et al 2003]。TreacleはrDNA転写物、核小体糸やタンパク輸送、核小体と細胞質の間のリボソームサブユニットに関与する[Winokur & Shiang 1998, Dauwerse et al 2011]。また、神経堤細胞遊走との関与も考えられている [Sakai & Trainor 2009]。
異常遺伝子産物
TCOF1遺伝子変異はTreacleタンパクのハプロ不全を引き起こす [Isaac et al 2000]。変異の大多数は転写の途中で終止コドンが導入され、ナンセンス変異依存のRNA分解機 の結果として、異常な遺伝子からのRNA転写物は失われる 。 このことが 罹患者の組織において異常な遺伝子とハプロ不全による タンパク産物の喪失につながる 。異常なタンパク産物をもたらすミスセンス変異はN末端あるいはC末端核移行シグナルを破壊し、第一および第二鰓弓発生時に 核へ輸送するタンパク質の 能力に 影響を及ぼし 、胚発生時に神経堤 細胞のアポトーシスを引き起こす [Marsh et al 1998, Jones et al 1999, Dixon et al 2000, Isaac et al 2000]。
POLR1C
遺伝子構造
POLR1C遺伝子は9個のエクソンから構成され、2つのイソ型をもつ。最も長い転写物は88bpの5'非翻訳領域と226bpの3'非翻訳領域をもつ1,355個のヌクレオチドの翻訳領域を含む。より詳細な遺伝子およびタンパクの情報はTable A、遺伝子記号を参照する。
病的アレル変異
6個のPOLR1C遺伝子へテロ接合変異はTCOF1遺伝子変異のない3名の患者に同定された[Dauwerse et al 2011]。ナンセンス変異、ミスセンス変異、スプライシングサイト変異、重複、挿入および欠失が含まれる [Dauwerse et al 2011]。
正常遺伝子産物
RNAポリメラーゼ1ポリペプチドDとCはそれぞれ16kd(133個のアミノ酸)と39kd(346個のアミノ酸)である。これらのサブユニットはRNAポリメラーゼIとRNAポリメラーゼIIIに働き、両方ともリボソームRNAの転写に関与する [Dauwerse et al 2011]。
異常遺伝子産物
POLR1C遺伝子へテロ接合変異はPOLR1Cタンパクの機能喪失 を引き起こす [Dauwerse et al 2011]。
POLR1D
遺伝子構造
POLR1D遺伝子は三つのエクソンから構成され、2つのイソ型をもつ。最も長い転写物118bp 5'非翻訳領域と1,458bp 3'非翻訳領域をもつ1,945個のヌクレオチドの翻訳領域を含む。より詳細な遺伝子およびタンパク情報はTable A、遺伝子記号を参照する。
病的アレル変異
20種類のPOLR1D遺伝子ヘテロ接合変異はTCOF1遺伝子変異を認めなかった患者に確認されている [Dauwerse et al 2011]。ナンセンス変異、ミスセンス変異、スプライシングサイト変異、重複、挿入と欠失が含まれる[Dauwerse et al 2011]。
正常遺伝子産物
RNAポリメラーゼ1ポリペプチドDとCはそれぞれ16kd(133個のアミノ酸)と39kd(346個アミノ酸)である。これらのサブユニットはRNAポリメラーゼIとRNAポリメラーゼIIIに働き、両方ともリボソームRNAの転写に関与する[Dauwerse et al 2011]。
異常遺伝子産物
POLR1D遺伝子変異はRNAポリメラーゼ1ポリペプチドDのハプロ不全を引き起こす[Dauwerse et al 2011]。
更新履歴
-
Gene Reviews著者: Sara Huston Katsanis, MS and Ethylin Wang Jabs, MD
日本語訳者: 江田 肖(瀬戸病院 遺伝診療科),石川亜貴(札幌医科大学医学部遺伝医学)
Gene Reviews 最終更新日: 2012.8.30 語訳最終更新日: 2015.1.6 (in present)
![]()