コルネリア デ ランゲ症候群
(Cornelia de Lange Syndrome)
[Synonyms: BDLS、ブラッハマン・デ ランゲ症候群、CdLS、デ ランゲ症候群]
GeneReviews著者: Matthew A Deardorff, MD, phD, Sarah E Noon, MS, and lan D Krantz, MD
日本語訳者:疋田美那子、升野光雄、山内泰子、黒木良和(川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
GeneReviews最終更新日: 2020.10.15. 日本語訳最終更新日: 2021.12.4.
要約
疾患の特徴
コルネリア デ ランゲ症候群(CdLS)は、軽度から重度まで様々な所見を有する。重症(古典的)CdLSは、特有の顔貌、発育不全(出生前発症:生涯を通じて5パーセンタイル未満)、多毛症、わずかな指節骨異常から乏指症(指の欠損)におよぶ上肢の減形成(減数異常)によって特徴づけられる。頭蓋顔面の特徴は、眉毛癒合、高い眉毛および/または濃い眉毛、長い睫毛、上向きの鼻孔を伴う短い鼻梁、歯間空隙の拡大した小さい歯、小頭症を含む。表現型が軽症である場合、成長や認知、四肢の障害への影響はそれほど深刻ではないが、CdLSと一致した特徴的な顔貌であることが多い。IQは30未満~102(平均:53)の範囲にある。多くは自閉的で自己破壊的な傾向を示す。しばしばみられる他の所見として、心中隔欠損症、胃腸機能障害、難聴、近視、停留精巣または性器発育不全が含まれる。
診断・検査
CdLSの診断は、示唆的な臨床所見のある発端者において、および/または分子遺伝学的検査によるNIPBL、RAD21、SMC3もしくはBRD4のヘテロ接合性病的バリアント、またはHDAC8もしくはSMC1Aのヘミ接合性病的バリアントの同定により確立する。
臨床的マネジメント
症状の治療:
胃腸回転異常の可能性の評価をし、胃食道逆流への積極的な管理を行う。逆流が重度の場合、噴門形成術を検討する。必要に応じて補充調整乳および/または胃瘻管造設により栄養必要量を満たす。精神運動発達やコミュニケーション能力を最適化するために、理学療法、作業療法、言語療法を行う。てんかん、視力障害、鼻涙管閉塞、難聴、口蓋裂、歯の形成および/または歯列異常、心疾患、停留精巣/尿道下裂、双角子宮、膀胱尿管逆流、貧血および/または血小板減少症、免疫不全には標準治療を行う。手術を検討する場合は、悪性高熱症の予防措置と血小板減少症や心疾患に対する術前評価を行い、麻酔下における気道の観察が推奨される。
経過観察:
診察時毎回:身体計測、栄養状態や安全な経口摂取の評価、胃食道逆流症(GERD)の徴候や症状、呼吸不全を伴う誤嚥の有無の観察、痙攣や自律神経機能障害の徴候など、新たにみられる症状の評価、成長発達や教育上のニーズの観察、不安、注意力、攻撃的な行動や自傷行為などの行動評価、運動能力や自立能力の評価を行う。少なくとも年1回:眼科評価、歯科評価と歯科クリーニング、小児期および青年期における聴力評価を行う。
遺伝カウンセリング
NIPBL遺伝子関連のCdLS、RAD21遺伝子関連のCdLS、SMC3遺伝子関連のCdLSおよびBRD4遺伝子関連のCdLSは常染色体優性遺伝形式である。HDAC8遺伝子関連のCdLSおよびSMC1A遺伝子関連のCdLSはX連鎖遺伝形式である。罹患者の大多数は、NIPBL遺伝子の新生のヘテロ接合性病的バリアントを有する。常染色体優性CdLSの1%に満たない人が、罹患した片親を持つ。両親が臨床的な症状を呈していない場合、生殖細胞系列モザイクの可能性があり、CdLSを持つ発端者の同胞へのリスクは1.5%と推定される。X連鎖の CdLS を持つ男性発端者の同胞へのリスクは、発端者の母親の遺伝的状況に依存する。X連鎖のCdLS を持つ女性発端者の同胞へのリスクは、発端者の母親と父親の遺伝的状況に依存する。リスクの高い妊娠に対する出生前検査と着床前遺伝学的検査は、病的バリアントが同定された家系では可能である。
診断
コルネリア デ ランゲ症候群(CdLS)は臨床的スペクトラムを構成し、より軽度の特徴を有する人から、より重度の古典的な特徴を有する人がいる。国際合意声明は、臨床遺伝学的検査の決定を支援するため、古典的と非古典的CdLSを定義する採点システムのみならず、基本的特徴と示唆的特徴の両方を定義している [Kline et al 2018]。
臨床診断
- 特有の頭蓋顔面の外観(しばしば認識可能;図1を参照)は以下を含む。
- 小頭症(平均頭囲:後頭前頭を結ぶ周囲径は2パーセンタイル未満)
- 高い眉毛および/または濃い眉毛を伴う眉毛癒合
- 長く濃い睫毛
- 短い鼻梁、上向きの鼻孔を伴う上向きの鼻尖
- 長く平坦な人中と上口唇の薄い唇紅、下向きの口角
- 口蓋裂の有無にかかわらず、高度な高口蓋
- 小さな歯、歯間空隙の拡大
- 下顎骨棘の有無にかかわらず、小顎症
- 成長障害は、出生前より指摘され、出生後も持続する可能性がある
- 発達遅滞/知的障害は軽度から重度まで様々
- 四肢の異常(対称性または非対称性のいずれか)は、前腕の完全な欠如を伴う重度の減形成(減数異常)から、多くは上肢を含む様々な形態の乏指症(指の欠損)、軽度では、小さな手(小肢症)および指節骨異常(第5指彎指症、短い第1中手骨による母指の近位位置異常)に至るまで、多岐にわたる
- 多毛症.濃い頭髪はしばしば側頭部へ広がり、時に顔、耳、背中、腕に広がる
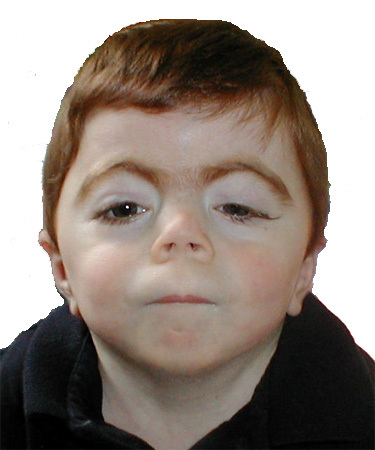
図1.
古典的CdLSの頭蓋顔面像
X線写真所見.四肢欠損のない患者では、単純X線写真で、母指の近位位置異常をきたす第1中手骨短縮の存在は、診断に有用である
診断の確定
CdLSの診断は、上記の臨床的特徴を有する発端者において、および/または分子遺伝学的検査により、NIPBL遺伝子、RAD21遺伝子、SMC3遺伝子または BRD4遺伝子のヘテロ接合性病的バリアントの同定、もしくは HDAC8 遺伝子または SMC1A 遺伝子のヘミ接合性病的バリアントが同定されることにより確立される(表 1 参照)。
分子遺伝学的検査のアプローチは、表現型に応じて、標的遺伝子検査(一連の単一遺伝子検査または多遺伝子パネル)と網羅的ゲノム検査(エクソームシークエンス解析、エクソームアレイ、ゲノムシークエンス解析)を組み合わせることができる。
標的遺伝子検査では、臨床医がどの遺伝子が関与している可能性が高いかを判断する必要があるが、ゲノム検査においてはそうではない。コルネリア デ ランゲ症候群の表現型は広範であるため、示唆的所見に記載されている顕著な特徴を持つ人は、標的遺伝子検査を用いて診断される可能性が高く(オプション1参照)、コルネリア デ ランゲ症候群の診断が考慮されていない人は、ゲノム検査を用いて診断される可能性がより高い(オプション2参照)。
オプション1
表現型がコルネリア デ ランゲ症候群の診断を示唆する場合、分子遺伝学的検査のアプローチは、多遺伝子パネルまたは一連の単一遺伝子検査の使用を含む。
コルネリア デ ランゲ症候群多遺伝子パネル.少なくともNIPBL、SMC1A、HDAC8、SMC3、RAD21およびBRD4並びに、AFF4、ANKRD11、CREBBPおよびEP300など、CdLSに類似した表現型を引き起こす可能性のあるいくつかの追加遺伝子(鑑別診断を参照)を含む、コルネリア デ ランゲ症候群の多遺伝子パネルは、原因となるバリアントを検出するために最も効果的な方法である [Kline et al 2018]。体細胞モザイクの頻度が高いため [Huisman et al 2013]、次世代シークエンス解析(NGS)などのモザイクを検出できる検査を検討すべきであり、できれば未培養の線維芽細胞を使用するか、口腔細胞または膀胱上皮細胞も使用できる。
このアプローチは、一方で意義不明なバリアントや根本的な表現型を説明できない遺伝子の病的バリアントの同定を制限しつつ、最も手頃な費用で疾患の遺伝学的原因を同定する可能性が高い。注釈:(1)パネルに含まれる遺伝子や各遺伝子に用いられる検査の診断感度は検査機関により様々で、時間とともに変化する可能性がある。(2) 一部の多遺伝子パネルには、このGeneReviewで論じられる疾患とは関連しない遺伝子を含むこともある。(3) 一部の検査機関では、パネルのオプションとして、検査機関ごとにカスタマイズされたパネルおよび/または臨床医によって指定された遺伝子を含む、表現型に焦点を合わせてカスタマイズされたエクソーム解析を使用できることがある。(4) パネルに用いられる方法には、シークエンス解析、欠失/重複解析および/またはその他のシークエンスに基づかない検査を含むことがある。CdLS については、欠失・重複解析も含む多遺伝子パネルが推奨される(表 1を参照)。
多遺伝子パネルの導入についてはこちらをクリック。遺伝学的検査を依頼する臨床医のためのより詳細な情報についてはこちらで見られる。
一連の単一遺伝子検査.NIPBL遺伝子、SMC1A遺伝子、HDAC8遺伝子、SMC3遺伝子、RAD21遺伝子およびBRD4遺伝子の分子遺伝学的検査を順次検討する。
- 国際的な合意採点基準において高いスコアでCdLS を強く示唆する特徴を持つ人や、多遺伝子パネル検査が利用できない人に対しては、まず NIPBL遺伝子のシークエンス解析が考慮される。病的バリアントが同定されない場合は、次にNIPBL遺伝子の標的遺伝子欠失/重複解析を考えるべきである。
- NIPBL遺伝子の病的バリアントが同定されず、罹患者の身体的特徴が軽度のCdLSの場合は、次にSMC1A遺伝子のシークエンス解析と標的遺伝子欠失/重複解析を検討しなさい。
- NIPBL遺伝子またはSMC1A遺伝子の病的バリアントが同定されず、CdLSが強く疑われる場合(特に軽度の特徴を持つ人では)、BRD4遺伝子、SMC3遺伝子、RAD21遺伝子とHDAC8遺伝子のシークエンス解析および標的遺伝子欠失/重複解析を検討すべきである。
オプション2
非典型的な表現型の特徴からコルネリア デ ランゲ症候群の診断が強くは考慮されない場合、網羅的なゲノム検査(どの遺伝子が関与している可能性が高いかについて臨床医が判断する必要はない)が最良の選択肢である。エクソームシークエンス解析が最も一般的に用いられている。ゲノムシークエンス解析も可能である。
網羅的ゲノム検査の導入についてはこちらをクリック。ゲノム検査を依頼する臨床医のためのより詳細な情報についてはこちらで見られる。
表1.
コルネリア デ ランゲ症候群に用いられる分子遺伝学的検査
| 遺伝子1, 2 | その遺伝子の病的バリアントに起因するCdLSの割合 | その方法によって検出できる病的バリアントを持つ発端者の割合3 | |
|---|---|---|---|
| シークエンス解析4 | 標的遺伝子欠失重複解析5 | ||
| BRD4 | 1%未満 | 3/4 6 | 1/4 6 |
| HDAC8 | 約4% | 約90% 7 | 約10% 7 |
| NIPBL | 約80% 8,9 | 約97% 9 | 約3% 10,11 |
| RAD21 | 1%未満 | 20/22 12 | 2/22 12 |
| SMC1A | 約5% 13 | 約100% | |
| SMC3 | 1%-2% 14 | 約97% | 約3% |
| 不明 | 3%-5% | ||
- このリストにある遺伝子はアルファベット順である。
- 染色体座およびタンパク質については、表A.遺伝子とデータベースを参照。
- これらの遺伝子で認められるアレルバリアントに関する情報は分子遺伝学を参照。
- シークエンス解析は、良性、おそらく良性、意義不明、おそらく病的または病的バリアントを同定する。バリアントには小さな遺伝子内欠失/挿入やミスセンスバリアント、ナンセンスバリアント、スプライス部位バリアントがある。一般的には、エクソンまたは全遺伝子の欠失/重複は同定されない。シークエンス解析結果の解釈について考慮すべき問題はこちらをクリック。
- 標的遺伝子の欠失/重複解析では、遺伝子内の欠失または重複を検出する。検査方法には、定量PCR、ロングレンジPCR、MLPA(multiplex ligation-dependent probe amplification)法、単一のエクソン欠失や重複の検出を目的とする標的遺伝子マイクロアレイがある。標的遺伝子の欠失/重複検査は、単一エクソンから遺伝子全体に至るまでの欠失を検出する。しかし、大きな欠失および/または隣接遺伝子の欠失(例えば、Olleyら[2018]によって記載されたもの)の切断点は、これらの方法によっては検出されない可能性がある。
- Olley et al [2018]
- Ansari et al [2014]、Kaiser et al [2014]。
- Borck et al [2004]、Gillis et al [2004]、Tonkin et al [2004]、Bhuiyan et al [2006]、Musio et al [2006]、Yan et al [2006]、Selicorni et al [2007]、Huisman et al [2013]
- NIPBL 遺伝子の体細胞モザイクは、約10%~15%の人に報告されている。末梢血検体の採血時に口腔検体の採取も考慮される[Huisman et al 2013]。CdLSが強く疑われ、末梢血検体の分子遺伝学的検査が正常であれば、NIPBL遺伝子モザイクのスクリーニングが求められる。
- NIPBL遺伝子の標的遺伝子の欠失/重複解析は、NIPBL-CdLSの約3%を検出する[Pehlivan et al 2012、Russo et al 2012、Ansari et al 2014]。
- NIPBL遺伝子を含む5p13の大きな欠失を持つ少数の報告例があるため、古典的なCdLSの特徴を持ちながらも分子遺伝学的検査が正常である場合には、細胞遺伝学的検査や染色体マイクロアレイ(CMA)も考慮される [Taylor & Josifek 1981、Hulinsky et al 2005、Hayashi et al 2007]。
- Deardorff et al[2012]、Krab et al[2020]
- Musio et al [2006]、Borck et al [2007]、Deardorff et al [2007]、Huisman et al [2017]。
- Gil-Rodríguez et al [2015]
臨床的特徴
臨床像
古典的コルネリア デ ランゲ症候群(CdLS)は70年以上前に正式に特徴づけられ、臨床的に非常に詳細に記述された [Ptacek et al 1963、Jackson et al 1993]が、CdLSの分子遺伝学的基盤が明らかになったことで、より軽度の特徴や非定型の特徴を持つ罹患者が認識されるようになってきた。したがって、この疾患は軽度から重度までの一連の所見を呈する(遺伝子による表現型相関を参照)。古典型CdLS より臨床的にはあまり顕著ではない軽度の表現型を持つ人が、CdLS を持つ人の大多数を占めているかもしれない [Deardorff et al 2007、Rohatgi et al 2010]。
表2.
コルネリア デ ランゲ症候群の特徴
| 特徴 | 特徴のある人の割合(%) | コメント |
|---|---|---|
| 眉毛癒合 | 98% 1 | 古典的特徴を持つ人において |
| 摂食困難 | >95% | |
| 発育不全 | >95% | 出生前に指摘されることもある |
| 知的障害 | >95% | 古典的なCdLSを持つ人では通常は重度から最重度である |
| 小さい手足 | >90% | |
| 小頭症・短頭 | >90% | |
| 長い睫毛 | >90% | |
| 上口唇の薄い唇紅縁 | >90% | |
| 下向きの口角 | >90% | |
| 歯の問題 | >90% | 永久歯萌出の遅れ、小さく欠損した歯、歯の転位、叢生歯、GERDによる齲歯、歯周病や歯ぎしり |
| 多毛症 | >80% | 顔、耳、背中、腕に及ぶ |
| 聴覚障害 | 80% | 40%以上で最重度のSNHLを伴うが、時間の経過とともに改善する可能性がある。 |
| 小顎症 | 80% | 古典的特徴を持つ人において |
| 橈骨頭の発育不全 | 79% | |
| 胃食道逆流症 | 75% | |
| 彎指趾症 | 70% | |
| 鼻涙管閉塞症 | 70% | |
| 眼瞼下垂 | 60% | |
| 大理石様皮膚 | 60% | |
| 自傷行為 | 56% | |
| 睡眠障害 | 50% | |
| 下顎骨棘 | 42% | |
| 側弯症 | 33% | |
| 高口蓋、口蓋裂 | 30% | |
| 先天性心疾患 | 30% | 最もよくあるのは肺動脈弁狭窄または末梢肺動脈狭窄、VSDとASD |
| 痙攣 | 25% | |
| 乏指症 | 25% |
特徴および頻度の大半は、Jackson et al [1993]、Kline at al [2018]とその中の参照文献による。
ASD=心房中隔欠損症;GERD=胃食道逆流症;SNHL=感音性難聴;VSD=心室中隔欠損症
成長.CdLSを持つ人のほとんどで出生前の成長障害が生じる。均整のとれた低身長をきたす対称性の緩徐な成長は、生後6か月までにより顕著となる。古典的なCdLSを持つ人では、平均身長と平均体重は生涯を通じて5パーセンタイル以下である [Boog et al 1999]。全体的に軽度の臨床的特徴および/またはモザイクの病的バリアントを持つ人では、成長への影響はそれほど深刻ではない。さらに、発育不全は、胃食道逆流症や摂食に関する他の問題(下記の胃腸を参照)に起因する二次的な体質的な発育不全が付加される可能性がある。
CdLS 特有の成長曲線が作成されている。www.cdlsusa.org(女子、男子)を参照。
知的障害.CdLSを持つ人の大多数に発達遅滞がある。IQレベルの範囲は30以下から102までと幅広く、平均IQは53である [Kline et al 1993、Saal et al 1993]。古典型の特徴を有する患者は、重度から最重度の知的障害を有する可能性が高い(遺伝型と表現型の相関を参照)。
- 表出言語は受容言語よりも損なわれることが多く、受容言語は認知よりも損なわれる [Ajmone et al 2014]。
- 効果的な言語的または非言語的コミュニケーション能力は、生活の質を非常に向上させる [Kline et al 2018]。早期の積極的・代替コミュニケーションの介入は非常に効果的である[Ajmone et al 2014]。
- 24か月までに半数の子どもが歩き、10歳までに95%の子どもが歩く。3歳までに半数の子どもが自分で食事摂取ができ、10歳までに95%の子どもが自分で食事摂取ができる [Kline et al 1993] 。
行動.様々な行動問題が報告されている [Kline et al 2018]。
- 行動の問題は多くの場合、意思疎通の困難さからの欲求不満に直接関連する(上記の知的障害を参照)。
- 感覚処理障害のため、(感覚刺激により)低感受性、過敏性、見当識障害、および固視を引き起こす可能性がある。
- 自閉的行動は、社会的相互作用や身体的な接触の回避もしくは拒絶につながる。
- 反復行動はよく見られ、不安により増悪することがあり、より顕著な知的障害や自閉的特徴と関連する。
- 臨床的に重大な自傷行為は56%の人に見られ、多くは手を使って自傷行為を行う。
神経.概してCdLSを持つ人の約25%は痙攣がある。部分てんかんは最も一般的なタイプで、通常2歳前に発症する。多くの患者は標準的な薬物療法が効果的であり、数年のちには薬物療法を中止することができる [Verrotti et al 2013] (臨床的マネジメント、症状の治療を参照)。
四肢合併症.上肢の重度の異常は CdLSを持つ人の25%に見られる。
- 上肢欠損は、前腕が完全にない重度の減形成(減数異常)から、様々な形態の乏指症(指の欠損)に至るまで、古典的な特徴を持つ人の約3分の1に見られる。
- 四肢欠損がない場合、ほぼすべての人に小肢症(小さな手)、母指の近位位置異常や第5指彎指症が見られる(図3を参照)。
- 橈尺骨癒合症はよく見られ、肘の屈曲拘縮をきたすことがある
- 下肢は上肢より影響は少ない。足はしばしば小さく、第2趾と第3趾の合指症が80%以上の罹患者に見られる [Jackson et al 1993]。
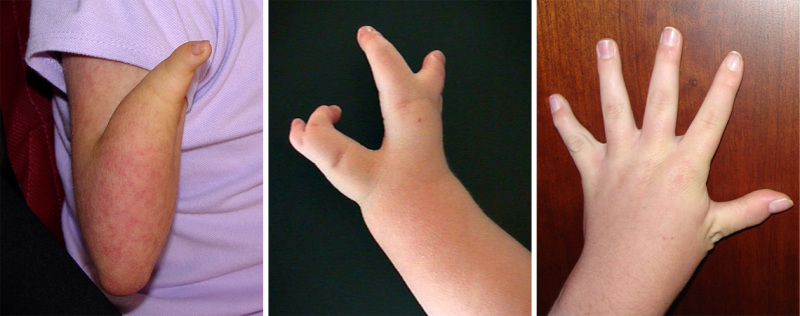
図3.
CdLSにおける四肢異常の範囲
胃腸.胃食道逆流症(Gastroesophageal reflux disease: GERD)は大多数の罹患者にみられる。食道炎、誤嚥、化学物質による肺炎、易刺激性といったGERDの他の合併症は、新生児期のGERDの診断と治療により回避できる(臨床的マネジメント参照)。約3分の1は誤嚥の既往を認め、15%は経管栄養を必要とする [Luzzani et al 2003]。その他の消化管の異常は以下の通りである。
- 新生児期の持続する嘔吐の原因として最も多い幽門狭窄症(4%)
- 腸回転異常(2%)
- 先天性横隔膜ヘルニア(CDH) (1%)
CDHは出生前と出生後にも診断されるが、特に周産期死亡した児において過小評価されているかもしれない。
眼科.罹患者の60%もが、近視(60%)と眼振(37%)を含む他の眼の問題だけでなく、ある程度の眼瞼下垂を示す [Levin et al 1990]。その他の眼科の異常は、
- 鼻涙管狭窄
- 緑内障
- 小角膜
- 乱視
- 視神経萎縮
- 視神経欠損
- 斜視
- 眼球突出
耳鼻咽喉科.感音性難聴はCdLSを持つ子供の40%にみられ、伝音性難聴は60%にみられる [Marchisio et al 2014]。注意すべきことは、多くの難聴(感音性難聴と伝音性難聴のどちらも)が時間の経過とともに改善することである [Janek et al 2016]。
腎尿路生殖器.腎異常は、主に膀胱尿管逆流で12%に報告されている。
- CdLSを持つ男性の73%に停留精巣が見られる
- 低形成の(小さい)性器は57%に見られ、男性の9%に尿道下裂がみられる [Jackson et al 1993] 。
- 20人の罹患女性のうち5人(25%)に腹痛の原因となり得る双角子宮が見られた [Oliver et al 2010、Kline et al 2015]。
心血管.CdLSを持つ人のおよそ30%は、先天性心疾患がある [Chatfield et al 2012]。最もよくある異常は以下の通りである(頻度が高いものから順に)
- 肺動脈弁狭窄または末梢肺動脈狭窄
- 心室中隔欠損症
- 心房中隔欠損症
- 大動脈弓縮窄または低形成
- 大動脈弁異常
- ファロー四徴症
- 両大血管右室起始症
- 共通房室弁口
免疫学.CdLSを持つ数名に抗体欠損が報告されており、重症または反復感染症罹患者において免疫学的スクリーニングや免疫不全症の管理の必要性を示している [Jyonouchi et al 2013]。最も多く報告されている反復感染症には、慢性耳感染症、慢性ウイルス性呼吸器感染症および肺炎が含まれる。T細胞機能障害は、CdLSを持つ人にみられる抗体欠損に関連している可能性がある。
血液学.血小板減少症は乳児期以降に消失することが多いが、まれに持続性の特発性血小板減少性紫斑病に移行する [Lambert et al 2011]。ほとんどの場合、貧血は一過性である。
その他の特徴
- 幼児期の終わりに消失傾向にある特徴的な低音域の泣き声は、CdLSを持つ子どもの75%に記述され、より重篤な症例と関連している[Jackson et al 1993]。
- 低形成の乳頭と臍は、50%に見られる。
予後.家族性症例の多くは、家系内で表現度が比較的一致している。臓器に重度の先天異常がない場合、平均余命は有意に低下しない。
- BeckとFenger [1985] は、1917年から 1982年の間に生まれた CdLS を持つ 48 人の死亡率を調査し、累積生存率を比較したところ、一般集団と比較して死亡率がわずかな増加を認めた。増加はより若い年齢群でより顕著であった。また、54歳と61歳で死亡した2人を報告した。
- Schrierら[2011]は、死因が判明している295人の罹患者を報告した。誤嚥/逆流および肺炎を含む呼吸器の原因が最も多い主因(31%)であり、次いで閉塞/軸捻を含む胃腸疾患(19%)が続いた。先天異常は死亡の15%を占め、先天性横隔膜ヘルニアと先天性心疾患が含まれる。後天性心疾患は死亡の3%を占めた。死亡原因は、神経学的原因と事故がそれぞれ8%、敗血症4%、癌2%、腎疾患1.7%、その他が9%を占めた。
遺伝子による表現型相関
NIPBL
特徴的顔貌や四肢の異常など、古典的なCdLSの所見を持つ人は、NIPBLの病的バリアントを持つ可能性が高い。
その他の遺伝子
NIPBLの病的バリアントを持つ人と比べると、特徴的顔貌の一部を有し、様々な認知機能や四肢または構造的障害を伴う、より軽度の表現型が一貫して記述されてきており、BRD4遺伝子、SMC3遺伝子もしくはRAD21遺伝子のヘテロ接合性病的バリアントを持つ人、またはHDAC8遺伝子もしくはSMC1A遺伝子のヘミ接合性病的バリアントを持つ人に見られる[Deardorff et al 2007、Selicorni et al 2007、Rohatgi et al 2010、Kaiser et al 2014、Minor et al 2014、Gil-Rodríguez et al 2015、Olley et al 2018](図2参照)。
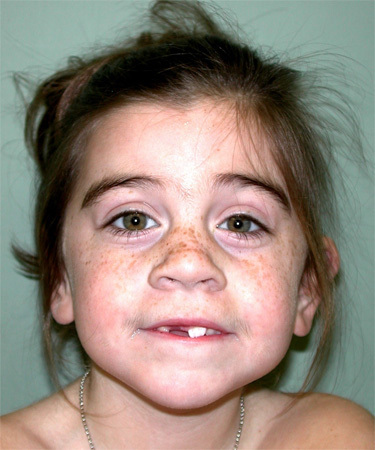
図2.
SMC1Aの病的バリアントを持つ罹患者
SMC1AおよびSMC3
- SMC1A遺伝子またはSMC3遺伝子の病的バリアントを持つ人は、典型的にはNIPBL遺伝子の病的バリアントを持つ人よりも構造異常が少なく、発育不全は軽度であるが、中等度から重度に及ぶ顕著な知的障害を有する [Deardorff et al 2007、Gil-Rodríguez et al 2015、Huisman et al 2017]。
- SMC1A遺伝子またはSMC3遺伝子の病的バリアントを持つ人の顔貌は、NIPBL遺伝子のヘテロ接合性病的バリアントを持つ人より、わずかであるがより平坦で幅の広い眉毛と、より広く長い鼻梁を持つ [Rohatgi et al 2010]。
- SMC3遺伝子の病的バリアントを持つ人は、特にしばしば眉毛癒合はわずかか欠如し、より広い球状の鼻、および長く整った人中を持つ。
- 心形態異常は、通常は軽度であるが、SMC3遺伝子の病的バリアントを持つ人でも観察され(約56%)、SMC1A遺伝子の病的バリアントを持つ人ではより頻度は低い [Gil-Rodríguez et al 2015]。
RAD21.
ヘテロ接合性RAD21遺伝子の病的バリアントを持つ人
- 通常は主要な構造上の特徴は見られない。
- 古典的なCdLSを持つ人と比較して軽度の認知機能障害を持つ。
具体的に、10%は正常な認知、45%は軽度の認知機能障害があり、重度または最重度の認知機能障害はない [Krab et al 2020]。
- 多くの場合、成長障害、軽度の骨格異常、およびCdLSと重なる顔貌が見られる [Deardorff et al 2012、Krab et al 2020]。
HDAC8
- HDAC8遺伝子のヘミ接合性の病的バリアントを持つ男性は、顔貌はCdLSと重複するが、通常は大泉門閉鎖遅延、眼瞼下垂、眼間開離、広い鼻、モザイク状の皮膚の色素沈着、歯の異常、朗らかで友好的な性格を示す。成長もまた、出生後の発育不全や小頭症の頻度は低く、それほど深刻な影響を受けない傾向がある。
- 女性:HDAC8遺伝子のヘテロ接合性病的バリアントを持つ女性の臨床症状は男性と重複するが、重症度はX染色体不活化パターンに大きく影響される [Kaiser et al 2014]。
遺伝型と表現型の相関
NIPBL.NIPBL遺伝子の病的ミスセンスバリアントおよびインフレーム欠失を持つ人は、NIPBL遺伝子の機能喪失型病的バリアントを持つ人と比較して、成長障害はあまり重度ではなく、知的障害は軽度で、構造的異常は少ないことが知られている [Gillis et al 2004、Ajmone et al 2014、Ansari et al 2014]。
SMC1A.CdLS表現型を引き起こす病的バリアントは、典型的にはミスセンスであり、重症度の幅の原因となる。注目すべきは、SMC1Aにおける機能喪失型病的バリアントは、早期乳児てんかん性脳症を引き起こす(OMIM 301044;遺伝学的に関連する(アレル)疾患を参照)。
命名法
コルネリア デ ランゲ症候群 (CdLS)は、1849年にVrolikによって最初に記述され、乏指(趾)症の極端な例として1例報告された [Oostra et al 1994]。Brachmann [1916]は、対称性の単指症、前肘部みずかき形成、小人症、頚肋骨と多毛症の1症例を詳細に記述した。
1930年代には、オランダの小児科医であるCornelia de Langeが、類似した特徴を持つ血縁ではない2人の少女について述べ、彼女が勤務していた都市にちなんで、この疾患をtypus degenerativus amstelodamensisと名付けた [de Lange 1933、de Knecht-van Eekelen & Hennekam 1994]。いくつかの文献では、この疾患をBrachmann-de Lange syndromeと呼んでいるが、デ ランゲ博士の疾患の理解への貢献に敬意を表して、Cornelia de Lange syndromeと広く呼ばれている。
有病率
CdLSの有病率は、軽度か様々な特徴を持つ人は気づかれない可能性があるため、推定することは困難である。公表された有病率の推定は、100,000に1人[Pearce & Pitt 1967]から10,000に1人 [Opitz 1985]にもわたる。EUROCATデータセットによるデータでは、古典型CdLS の有病率は 50,000に1人と推定される [Barisic et al 2008]。この数値には、より軽症でよくある表現型が含まれる可能性は少ない。
遺伝学的に関連する(アレル)疾患
このGeneReviewで論じられる、BRD4、HDAC8、NIPBLまたはSMC3の病的バリアントと関連する表現型以外の表現型は知られていない。
表4.
コルネリア デ ランゲ症候群の鑑別診断で考慮すべき遺伝子
| 遺伝子 | 疾患 | MOI | 鑑別疾患の特徴 | |
|---|---|---|---|---|
| CdLSとの重なり | CdLSとの識別 | |||
| AFF4 | CHOPS症候群 (OMIM 616368) |
AD | 認知機能障害、心疾患、低身長、骨格系異形成 | 粗な顔貌、肥満、 肺病変(CdLSでは一般的ではない特徴)1 |
| ANKRD112 | KBG症候群 | AD | DD/ID、低身長、濃い眉毛と眉毛癒合、上向きの鼻孔3 | IDは一般的により軽度。巨歯症、先天異常は少ない |
| ASXL1 | Bohring-Opitz症候群(BOS) | AD | DD/ID、出生前と生後の発育不全、小頭症、多毛症、小さな足、CdLSと類似の顔貌(特に乳児) | 栄養不耐性(栄養摂取が進まない) はより深刻である。強度近視、BOS姿勢4 、目立つ眼球 |
| EP300 | EP300 ルビンシュタイン・テイビ症候群(RSTS) |
AD | DD/ID、低身長、多毛症、濃い眉毛、長い睫毛、小顎症、回転異常5 | 突出した鼻と幅広い母指(どちらも CREBBP-RSTSよりもEP300-RSTSでは一般的ではない) 6 |
| TAF1 | 知的障害症候群 (OMIM 300966) |
XL | DD/ID、長い人中、上向きの鼻孔、小頭症、難聴7 | d width="141" >全身性筋緊張低下、長い顔、突出した耳|
| TAF6 | Alazami-Yuan症候群 (OMIM 617126) |
AR | 低身長、小頭症、彎指趾症、多毛症、DD/ID、長い人中、薄く弓状の眉毛、眉毛癒合を伴う顔貌8 | d width="141" >富士額(稀)|
AD=常染色体優性遺伝;AR=常染色体劣性遺伝;CdLS=コルネリア デ ランゲ症候群;DD=発達遅滞;ID=知的障害;MOI=遺伝形式;XL=X連鎖
- Izumi et al [2015]
- KBG症候群は、ANKRD11のヘテロ接合性病的バリアントまたはANKRD11を含む16q24.3の欠失のいずれかによって起こる
- Ansari et al [2014]、Parenti et al [2016]
- Bohring-Opitz症候群(BOS)を持つ人は、「BOS姿勢」と呼ばれる特定の四肢の姿勢を呈することがある。それは、手首の屈曲と中手指節関節での手指の屈曲を伴う肩の外旋か内転と記述される。
- Woods et al [2014]
- EP300の病的バリアントは、CREBBPの変異によって引き起こされるRSTSに似た表現型を引き起こす。しかし、EP300-RSTSでは、低くぶら下がった鼻柱を除いて、顔面の特徴はあまりみられない。母指趾は幅広いが、(橈側)偏位は非常にまれである。知的障害の程度は様々であるが、通常は比較的軽度であり、正常であることもある。(ルビンシュタイン・テイビ症候群を参照)。
- O'Rawe et al [2015]
- Alazami et al [2015]、Yuan et al [2015]
染色体2q31.1の欠失(OMIM 613681).HOXDクラスターを含むこの領域の欠失は、泌尿生殖器異常や発達異常だけでなく、CdLSで見られるのと同様に四肢の減形成(減数異常)ももたらす [Del Campo et al 1999]。染色体2q31.1の欠失を持つ人は、CdLSの特徴的顔貌は見られない。
Fryns症候群はCdLSの鑑別診断で考慮すべきで、どちらの疾患も、最も頻繁に横隔膜の異常(横隔膜ヘルニア、横隔膜挙上症、横隔膜低形成/無形成)が見られるからである。Fryns症候群とCdLSは、口蓋裂や長い人中、小顎症、遠位指低形成(爪、末端骨)だけでなく、心異常や腎異常によって特徴付けられる。しかし、Fryns症候群ではその他にも様々な特徴を有する(例えば、眼間開離、広い鼻梁や広い口)。Fryns症候群では新生児期以降の生存は稀であるが、一般的には重度の発達遅滞や知的障害を伴う。Fryns症候群の分子診断は、示唆的な所見と分子遺伝学的検査により特定されたPIGN遺伝子の両アレルに病的バリアントを持つ発端者において確立することができる。Fryns症候群の臨床診断を受けた人には、PIGN病的バリアントが同定されていないこともあるので、Fryns症候群の遺伝的異質性は依然として高い可能性がある。先天性横隔膜ヘルニアの概観も参照。
胎児性アルコール症候群(FAS).FASとCdLSの共通の特徴は、子宮内発育遅滞、発育不全、発達の異常、多動、小頭症、新生児の顔の多毛症、短い眼瞼裂、上向きの鼻を伴う短い鼻、長く平坦な人中、上口唇の薄い唇紅と心疾患を含む。しかし、FASでは手足は小さくなく、言葉はCdLSより影響を受けない。妊娠中のアルコール摂取の既往は、FASをCdLSと区別することに役立つ。
臨床的マネジメント
コルネリア デ ランゲ症候群の臨床管理ガイドラインが発表されている[Kline et al 2007、Kline et al 2018](全文)。
最初の診断後における評価
コルネリア デ ランゲ症候群と診断された人の疾患の程度とニーズを確かめるために、表5にまとめられた評価(診断に至る評価の一部として実施されていない場合)が推奨される。
表5.
コルネリア デ ランゲ症候群を持つ人の最初の診断後の推奨評価
| 系統/関心事 | 評価 | コメント |
|---|---|---|
| 体格 | 成長パラメータの測定 | CdLS専用の成長曲線が利用可能である1 |
| 神経 | 神経学的評価 | 痙攣が懸念される場合は脳波を考慮 |
| 発達 | 発達評価 |
|
| 行動 | 神経精神医学評価 | 12か月以上の人:睡眠障害、ADHD、不安および/またはASDを示唆する特性を含む行動の懸念についてスクリーニングする |
| 筋骨格 | 上肢X線写真を検討 | 橈尺骨癒合症の評価のために |
| 整形外科/物理療法学とリハビリテーション/PT/OTによる評価 | 以下の評価を含むために:
|
|
| 胃腸/摂食 | 消化器病学/栄養/摂食のチームによる評価 |
|
| 眼 | 眼科評価 | 鼻涙管開存性評価、眼瞼下垂、視力評価、散瞳眼底検査、眼圧測定 |
| 聴覚 | 聴覚評価2 | 難聴の評価 |
| 耳鼻咽喉科/口 | 口蓋裂の臨床評価 |
|
| 歯 | 歯科の評価 | 口蓋裂がある場合、または歯牙萌出後の乳児期に |
| 心血管 | 心エコー図 | 先天性心疾患の評価 |
| 泌尿生殖器 | 男性の停留精巣および/または尿道下裂の評価 | 泌尿器科医への紹介を検討 |
| 腎エコー検査 | 腎臓の構造的異常の評価 | |
| VCUGの検討 | 臨床的な必要に応じて膀胱尿管逆流の評価のために | |
| 骨盤部エコー検査の検討 | 思春期女性の子宮の異常のスクリーニング | |
| 血液 | 全血球計算 | 貧血、紫斑および/または出血の兆候がある場合 |
| 免疫 | 全血球計算と免疫プロファイル3 | 繰り返す感染が見られる場合、免疫学専門医への紹介を検討 |
| 多方面の/その他 | 臨床遺伝専門医および/または遺伝カウンセラーとの相談 | 遺伝カウンセリングを含むために |
| 家族への支援/社会資源 | 評価:
|
ASD=自閉症スペクトラム障害;GERD=胃食道逆流症;OT=作業療法;PT=理学療法;VCUG=膀胱尿管造影検査
- www.cdlsusa.org(女子、男子)
- 聴性脳幹反応検査(ABR)、耳音響放射検査を含む
- 定量的免疫グロブリン;破傷風、ジフテリア、肺炎球菌に対する抗体;B細胞像;T細胞像
症状の治療
表6.
コルネリア デ ランゲ症候群をもつ人の症状の治療
| 症状/関心事 | 治療 | 考慮すること/その他 |
|---|---|---|
| 体重増加不良/発育不全 | 摂食療法;摂食障害が持続する場合は胃瘻管造設を要することもある |
|
| 胃食道逆流症 |
|
重症の場合、噴門形成術を検討する |
| 回転異常 | 外科的修復 | |
| てんかん | 神経内科医によるAEDを用いた標準治療 |
|
| DD/ID | 発達遅滞/知的障害管理の問題を参照 | 言語療法の早期開始 |
| 四肢の欠損 | 腕/手の外科的介入を検討 整形外科/物理療法学とリハビリテーション/PT/OT |
必要になることは極めて稀 体位設定および移動補助具、障害者駐車用の掲示の必要性の検討 |
| 眼瞼下垂、斜視および/または視覚異常 | 眼科医が推奨する標準治療 | 地域の視覚サービスの早期介入または学区での対応 |
| 鼻涙管閉塞症 | 眼科医による積極的な治療 | 狭く変形した管のため、マッサージ治療は多くの場合効果的ではない |
| 聴覚 | 耳鼻咽喉科医による補聴器は有益である | 地域の聴覚サービスの早期介入または学区での対応 |
| 口蓋裂 | 可能であれば集学的頭蓋顔面チームによる標準治療 | |
| 歯の形成および/または位置の異常 | 歯科医および/または矯正歯科医による標準治療 | |
| 先天性心疾患 | 循環器科医による標準治療 | |
| 停留精巣/尿道下裂 | 泌尿器科医による標準治療 | |
| 膀胱尿管逆流 | ||
| 双角子宮 | 婦人科医による標準治療 | |
| 貧血および/または血小板減少症 | 血液内科医による評価 | 重度の血小板減少症では、IgG静脈内投与および/またはステロイド治療を必要とする場合がある |
| 免疫不全 | 免疫科医による標準治療 | 特別な懸念がない限り、定期的な予防接種を行う |
| 手術のリスク |
|
ミダゾラムに対する副作用と悪性高熱症がみられるが、まれである2(悪性高熱症感受性を参照) |
| 家族/地域 |
|
|
AED=抗てんかん薬;DD/ID=発達遅滞/知的障害;GERD=胃食道逆流症;OT=作業療法;PT=理学療法
- 一般的な痙攣の説明に関する両親/介護者への教育が適切である。てんかんと診断された子どものための非医学的介入や対処方法に関する情報は、「てんかんとわが子のためのツールキット」を参照のこと。
- Papadimos & Marco [2003]、Stevic et al [2015]、Moretto et al [2016]
発達遅滞/知的障害管理の問題
次の情報は、米国における発達遅滞/知的障害のある人に対する一般的な管理上の推奨事項を示している。標準的な推奨事項は国によって異なる場合がある。
0~3歳.乳幼児の精神保健サービスや特別支援教育者、感覚障害の専門家だけでなく、作業療法、理学療法、言語療法、摂食療法へのアクセスには、早期介入プログラムへの紹介が推奨される。米国では、早期介入はすべての州で利用可能な連邦政府出資のプログラムであり、個々の治療ニーズを対象とした在宅サービスを提供している。
3〜5歳.米国では、地元の公立学区を通じた発達幼稚園が推奨されている。入園前に、必要なサービスと療法を決定するための評価が行われ、確定された運動、言語、社会、認知機能の遅延に基づいて、適格とされた人達に対して個別教育計画(IEP)が展開される。通常、早期介入プログラムでは、この移行を支援する。発達幼稚園は通園型であるが、医学的に不安定で通園できない子どもたちのために、在宅サービスが提供される。
全年齢.発達小児科医との相談は、適切な地域、州および教育機関(米国)の関わりを確保し、生活の質を最大化する上で親を支援するために推奨される。考慮すべきいくつかの問題は:
- 個別教育計画(IEP)サービス:
- IEPは、サービスを受ける資格のある子どもたちに特別に考えられた指導と関連するサービスを提供する。
- IEP サービスは毎年見直され、変更が必要かどうかを判断する。
- 特別教育法で義務付けられているように、子どもたちは学校でできるだけ制限の少ない環境に置かれ、可能な限りで適切な場合に一般教育に組み込まれなければならない。
- 視覚と聴覚の相談員は、学習教材へのアクセスを支援するために、子どものIEPチームの一員に入るべきである。
- 理学療法、作業療法および言語療法は、子どもの学習教材へのアクセスに影響を与える上で必要な場合、IEPより提供される。それ以上は、罹患した個人のニーズに基づき、民間の支持療法が検討される場合がある。治療の種類に関する具体的な推奨は、発達小児科医により作成される。
- 子どもが10代に入ると、IEPで移行計画ついて話し合い、組み込まれる。IEPサービスを受けている場合、公立学区によるサービス提供は21歳になるまで義務付けられている。
- 504プラン(第504条:障害に基づく差別を禁止する米国連邦法規)は、教室で前の方の座席へ移動、(障害を持つ人の日常生活の)支援技術機器、授業で筆記する人、授業の合間の時間に余裕をもつ、修正された宿題や拡大されたテキストなどの適応や修正を必要とする人に考慮される。
- 発達障害管理(DDA)への登録を勧めている。DDAは、資格のある人にサービスとサポートを提供する米国の公的機関である。資格は州によって異なるが、通常は診断および/または関連する認知/適応障害によって決定される。
- 収入や資源が限られている家庭は、障害のある子どもの補足的保障所得(SSI)の資格を得ることもできる。
運動機能障害
粗大運動機能障害
- 可動性を最大にし、遅発性の整形外科的合併症(例:拘縮、脊椎側彎症、股関節脱臼)のリスクを減らすために、理学療法が推奨される。
- 必要に応じて、耐久性のある医療機器や体位設定機器の使用を考慮する(例:車椅子、歩行器、入浴用椅子、装具、適応型ベビーカー)。
- 筋緊張亢進やジストニアを含む筋緊張の異常に対して、バクロフェン、チザニジン、ボトックス®、抗パーキンソン薬または整形外科的処置の管理の助けとなる、適切な専門家の関与を検討する。
微細運動機能障害.摂食、身だしなみ、更衣、筆記などの適応機能に影響を与える微細運動能力が困難な場合、作業療法が推奨される。
口腔運動機能障害 は受診の度に評価し、食事中の窒息/嘔吐、体重増加不良、頻発する呼吸器疾患または他に説明のつかない摂食拒否が見られる場合には、臨床的な摂食評価および/またはX線検査による嚥下機能評価を受ける必要がある。子どもが安全に口から摂取できるとすれば、協調運動または感覚に関連した摂食の問題の改善を助けるために、通常は作業療法士または言語療法士による摂食療法が推奨される。食事は安全のために、とろみをつけたり、冷やしたりする。摂食機能障害が重度の場合は、経鼻胃管や胃管が必要となることもある。
コミュニケーション問題.表出言語の困難を持つ人に対して、代替コミュニケーション手段(例えば、拡大代替コミュニケーション [AAC])の評価を検討する。AACの評価は、その領域の専門知識のある言語聴覚士が行う。評価では、認知能力や感覚障害を考慮して、最も適切なコミュニケーション形態を決定する。AACに使用する機器には、画像交換コミュニケーションのような簡単な技術のものから、音声生成装置のような高性能なものまである。一般的な認識に反して、AAC機器は話し言葉の発達を妨げるものではなく、多くの場合、改善し得る。
社会的/行動的懸念
子どもは、応用行動分析(ABA)を含む自閉症スペクトラム障害の治療に使用される介入を受ける資格があり、その恩恵を受けることができる。ABA療法は、個々の子どもの行動的、社会的および適応的な長所と短所を対象としており、通常認定を受けた行動分析者と一対一で実施される。
発達小児科医との相談は、適切な行動管理戦略を通して両親を導き、必要に応じて注意欠陥多動障害の治療に使用される薬剤などの処方薬を提供するのに役立つ場合がある。
深刻な攻撃的行動や破壊的行動についての懸念は、小児精神科医により対処される。
経過観察
表7.
コルネリア デ ランゲ症候群をもつ人に推奨される経過観察
| 系統/関心事 | 評価 | 頻度 |
|---|---|---|
| 摂食 |
|
毎回の受診ごとに |
| 胃腸 | GERD徴候、症状の観察 | |
| 呼吸器 | 誤嚥の証拠、呼吸不全の観察 | |
| 神経 | 臨床適応があれば、痙攣がある人の観察 | |
| 痙攣や自律神経機能障害の徴候など、新たな症状の評価 | ||
| 発達 | 発達の向上と教育ニーズの観察 | |
| 行動 | 不安、注意、攻撃的もしくは自傷行為の行動評価 | |
| 筋骨格 | 物理療法学、OT/PTによる可動性の評価、自立能力 | |
| 多方面の/その他 | 家族に必要なソーシャルワーク支援(例:緩和ケア/レスパイトケア、訪問看護、その他の地域資源)とケアの調整 | |
| 眼科 | 眼科評価 | 少なくとも年1回 |
| 歯科 | 歯科評価とクリーニング | |
| 聴力 | 聴力評価 | 小児期・青年期は少なくとも年1回 |
GERD=胃食道逆流症;OT=作業療法;PT=理学療法
リスクのある血縁者の評価
遺伝カウンセリングの目的で、リスクにある血縁者の評価に関する問題については、遺伝カウンセリングを参照。
研究中の治療法
広い範囲の疾患と健康状態に対する臨床研究情報にアクセスするためには、米国のClinicalTrials.govおよび欧州のEU Clinical Trials Registerを検索のこと。注釈:この疾患における臨床試験はないかもしれない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
NIPBL関連のコルネリア デ ランゲ症候群(CdLS)、RAD21関連のCdLS、SMC3関連のCdLSおよびBRD4関連のCdLSは、一般に新生の病的バリアントによって引き起こされる常染色体優性疾患である。
HDAC8関連のCdLSおよびSMC1A関連のCdLSは、一般に新生の病的バリアントによって引き起こされるX連鎖疾患である。
家族構成員のリスク
常染色体優性遺伝
発端者の両親
- 常染色体優性遺伝のCdLSを持つ人の約99%は、新生の病的バリアントの結果として障害を持つ。
- CdLSと診断された人のうち罹患した片親がいるのは1%未満である。
- 孤発例(すなわち、家系に1人だけの発症)と思われる発端者の両親に推奨される評価には、成長パラメータの記入を完備したCdLS の特徴についての診察と、発端者に原因となる病的バリアントが同定されている場合の分子遺伝学的検査が含まれる。
- 発端者の病的バリアントがどちらの親の白血球DNA中にも検出されない場合、可能な説明は、発端者の新生の病的バリアントまたは片親の生殖細胞系列モザイクである。親の生殖細胞系列モザイクの頻度は約1.5%であるが、まれな家族例が非常に濃縮された集団では3.4%~5.4%の頻度が報告されている [Slavin et al 2012; 著者、未発表データ]。
発端者の同胞
発端者の同胞のリスクは、発端者の両親の遺伝的状況に依存する。
- 発端者の片親が罹患者および/または発端者に同定された病的バリアントを持つことが知られている場合、同胞の再発率は50%である。
- 発端者に見られるCdLS関連の病的バリアントがいずれの親の白血球DNAにも検出されない場合および/または両親が臨床的に罹患していない場合、生殖細胞系列モザイクの可能性があるため、発端者の同胞のリスクは1.5%と推定される [Jackson et al 1993]。
発端者の子
- 常染色体優性遺伝のCdLSを持つ人の子どもは、各々50%の確率で病的バリアントを受け継ぐ。
- CdLS の家系内再発の大多数は、表現型の正常な片親の生殖細胞系列モザイクの結果であるが、軽度の CdLS 罹患者が CdLS がある子どもを持つ稀な症例が報告されている。
他の家族構成員
他の家族構成員のリスクは発端者の両親の状況に依存する。もし、片親が罹患していれば、彼または彼女の家族構成員はリスクがあるだろう。
X連鎖遺伝
男性発端者の両親
- X連鎖のCdLSを持つ人のほとんどは、SMC1AまたはHDAC8の新生病的バリアントの結果として障害を持つ。
- 罹患男性の父親はCdLSを有さず、病的バリアントのヘミ接合性でもないであろう。
- 罹患者が複数人いる家族では、罹患男性の母親は通常は原因となるバリアントのヘテロ接合性である。代わりに、彼女が他に罹患した血縁者がいなくて、原因となるバリアントが白血球DNAに検出されないなら、彼女は生殖細胞系列モザイクをもつ可能性が最も高い。
- 注釈:SMC1A遺伝子は、典型的なX連鎖遺伝子と異なり、X染色体不活化の過程で完全には不活化が起こらない。この状況では、ヘテロ接合性の母親は、罹患した息子よりも軽度な CdLS の特徴を示す可能性が高い。しかし、これまでのところ、このモデルを十分に評価するには、SMC1A関連のCdLSを有する家族数があまりにも少ない。
女性発端者の両親
- 女性発端者は、母親か父親のいずれかから原因となるバリアントを受け継いでいるか、もしくは新生の病的バリアントによる。
- 両親の詳細な評価と広範囲の家族歴の検討により、発端者が新生の病的バリアントによるものか、病的バリアントを受け継いだことによるものかを区別するのに役立つ。両親の分子遺伝学的検査は、病的バリアントが遺伝したかどうかの判断につながる。注釈:親の白血球DNA検査では、体細胞モザイクのすべての例を検出することはできない。
- SMC1A病的バリアントに対する生殖細胞系列モザイクが、複数の罹患女性の父親において報告された [Deardorff et al 2007]。
男性発端者の同胞.
男性発端者の同胞のリスクは、母親の遺伝的状況に依存する:
- 発端者の母親がSMC1A遺伝子またはHDAC8遺伝子の病的バリアントを持つ場合、各妊娠でそのバリアントを伝える確率は50%である。
- 病的バリアントを受け継ぐ男性同胞は罹患する。
- 病的バリアントを受け継ぐ女性同胞はヘテロ接合性となる。SMC1A 遺伝子の病的バリアントのヘテロ接合性である女性は、CdLS のいくらかの特徴を有する可能性が高い。HDAC8 遺伝子の病的バリアントのヘテロ接合性である女性は、X連鎖不活化パターンにより、CdLS の臨床所見を呈する場合と呈さない場合がある。
- 発端者が孤発例(すなわち、家系に1人だけの発症)で、母親の白血球DNAから病的バリアントを検出できない場合、母親の生殖細胞系列モザイクの可能性があるため、同胞のリスクは約1%である。
女性発端者の同胞.
同胞のリスクは、両親の遺伝的状況に依存する:
- 発端者の母親がSMC1A遺伝子またはHDAC8遺伝子の病的バリアントを持つ場合、各妊娠で変異を伝える確率は50%である。この病的バリアントを受け継ぐ男性は罹患し、この病的バリアントを受け継ぐ女性はヘテロ接合体となる(男性発端者の同胞を参照)。
- 発端者の父親が病的バリアントを持つ場合、すべての娘にそのバリアントを伝えるが、息子には伝えない。
- 発端者が孤発例(すなわち、家系に1人だけの発症)で、どちらの親の白血球DNAからも病的バリアントが検出されない場合、親の生殖細胞系列モザイクの可能性があるため、同胞のリスクは約1%である。
発端者の子
- 重度のCdLSを持つ人は、通常は子を持たないが、軽症の罹患者は子をもうけることもある。
- X連鎖のCdLSを持つ男性は、病的バリアントを全ての娘に伝え、息子には伝えない。
- X連鎖のCdLSを持つ女性は、病的バリアントをそれぞれの子どもに50%の確率で伝える。
他の家族構成員.
他の家族構成員のリスクは、発端者の両親の状況に依存する:片親が病的バリアントを持つ場合、彼または彼女の家族構成員はリスクがあるだろう。
遺伝カウンセリングに関連した問題
家族計画
- 遺伝的リスクの決定と出生前/着床前遺伝学的検査の有用性について討議するための最適な時機は妊娠前である。
- 罹患している、もしくはリスクにある若年成人に遺伝カウンセリング(子への潜在的なリスクや生殖の選択肢についての討議を含む)を提供することは適切である。
DNAバンクは、(通常は白血球から抽出された)DNAを将来の使用のために保存しておくものである。検査法や遺伝子、アレルバリアントおよび疾患に対する我々の理解が、将来進歩する可能性があり、罹患者のDNA保存は考慮に値する。
出生前検査および着床前遺伝学的検査
分子遺伝学的検査.罹患家族構成員の中でCdLSの原因となる病的バリアントが同定されると、出生前検査や着床前遺伝学的検査が可能になる。
超音波検査.高解像度の超音波検査では、成長をフォローし、CdLSで影響を受ける四肢、心臓、横隔膜、口蓋、並びに他の臓器または構造の評価を行うことが、病的バリアントの確認されていない家族に提供できるだろう。出生前の超音波検査所見の報告として:
- 第1三半期での後頸部肥厚の増大 [Sekimoto et al 2000、Huang & Porto 2002、Clark et al 2012]
- 第2三半期で典型的となる成長障害
- 小顎症、目立つ上口唇とやや上向きの鼻孔を伴う平坦な鼻稜から成るCdLSを伴う胎児の子宮内の典型的な顔の輪郭 [Ranzini et al 1997、Boog et al 1999、Urban & Hartung 2001、Clark et al 2012] 。
母体血清スクリーニング.CdLSの胎児を妊娠している母体では、母体血清PAPP-A(pregnancy-associated plasma protein A)の値は、第1三半期および第2三半期において低くなるだろう [Westergaard et al 1983、Aitken et al 1999、Arbuzova et al 2003、Clark et al 2012]。
資源
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- CdLS World
CdLSワールドは、コルネリア デ ランゲ症候群で結ばれた世界中の組織やコミュニティのための国際的な「ハブ」である。国別の連絡先は、CdLSのウェブサイトで入手可能である。
www.cdlsworld.org
- Cornelia de Lange Syndrome Foundation, Inc.(コルネリア デ ランゲ症候群財団)
302 West Main Street
#100
Avon CT 06001
電話:800-223-8355(フリーダイヤルサポートライン)、860-676-8166
ファックス:860-676-8337
メール: info@cdlsusa.org
www.cdlsusa.org
- My46 Trait Profile
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A.
コルネリア デ ランゲ症候群:遺伝子およびデータベース
| 遺伝子 | 染色体座 | タンパク質 | Locus-Specific Datebases(座位特異的データベース) | HGMD | ClinVar |
|---|---|---|---|---|---|
| BRD4 | 19p13.12 | ブロモドメイン含有タンパク質4 | BRD4 | BRD4 | |
| HDAC8 | Xq13.1 | ヒストンデアセチラーゼ8 | HDAC8@LOVD | HDAC8 | HDAC8 |
| NIPBL | 5p13.2 | ニップドB様タンパク質 | NIPBL@LOVD | NIPBL | NIPBL |
| RAD21 | 8q24.11 | 二重鎖切断修復タンパク質rad21ホモログ | RAD21 | RAD21 | |
| SMC1A | Xp11.22 | 染色体タンパク質1Aの構造維持 | SMC1A@LOVD | SMC1A | SMC1A |
| SMC3 | 10q25.2 | 染色体タンパク質3の構造維持 | SMC3@LOVD | SMC3 | SMC3 |
データは以下の標準的な参考文献を編集したものである:HGNCによる遺伝子記号;OMIMによる染色体座、UniProtによるタンパク質名。リンクが提供されているデータベース(Locus Specific, HGMD, ClinVar)の記述については、ここをクリック。
表B.
OMIMに登録されているコルネリア デ ランゲ症候群(OMIMですべてを参照のこと)
| 22470 | CORNELIA DE LANGE SYNDROME 1; CDLS1 |
| 300040 | STRUCTURAL MAINTENANCE OF CHROMOSOMES 1A; SMC1A |
| 300269 | HISTONE DEACETYLASE 8; HDAC8 |
| 300590 | CORNELIA DE LANGE SYNDROME 2; CDLS2 |
| 300882 | CORNELIA DE LANGE SYNDROME 5; CDLS5 |
| 606062 | STRUCTURAL MAINTENANCE OF CHROMOSOMES 3; SMC3 |
| 606462 | RAD21 COHESIN COMPLEX COMPONENT; RAD21 |
| 608667 | NIPPED-B-LIKE; NIPBL |
| 608749 | BROMODOMAIN-CONTAINING PROTEIN 4; BRD4 |
| 610759 | CORNELIA DE LANGE SYNDROME 3 WITH OR WITHOUT MIDLINE BRAIN DEFECTS; CDLS3 |
| 614701 | CORNELIA DE LANGE SYNDROME 4 WITH OR WITHOUT MIDLINE BRAIN DEFECTS; CDLS4 |
分子学的病因論
コルネリア デ ランゲ症候群を引き起こす病的バリアントは、クロマチンを調節する遺伝子、最も一般的にはコヒーシン複合体を破壊する。コヒーシンは、染色体分離、ゲノムの安定性、ゲノムの構成、転写調節など、クロマチン生物学の多くの要素を調節するブレスレットのような構造体である。コヒーシン複合体のコアタンパク質には、SMC1A、SMC3、RAD21が含まれる。調節タンパク質のNIPBLとMAU2は、コヒーシンをクロマチンに導く際に重要な役割を果たし、HDAC8は、コヒーシンをクロマチンへ導き、安定化させるために重要である。かなり興味深いことに、プロモーターのエンハンサー制御を促進し、さらにRNAポリメラーゼ活性を促進するためのクロマチンのループ化を可能にするコヒーシンの重要な役割は、ANKRD11、EP300、BRD4およびAFF4によっても制御されており、これらの遺伝子の病的バリアントがCdLS様の表現型をどのようにしてもたらすかという点において、機能的に重複することが示唆される。このことから、CdLSは "コヒーシノパチー "および "トランスクリプトモパチー "と呼ばれている。
表 コルネリア デ ランゲ症候群:病気の原因のメカニズム
| 遺伝子1 | CdLS発症のメカニズム |
|---|---|
| BRD4 | 機能喪失 |
| HDAC8 | 機能喪失(タンパク質レベルの低下または酵素活性の低下) |
| NIPBL | 機能喪失 |
| RAD21 | 機能喪失 |
| SMC1A | 異常機能の獲得または優性阻害の可能性が高い |
| SMC3 | 異常機能の獲得または優性阻害の可能性が高い;非定型例ではまれに機能喪失 |
- 表1からの遺伝子はアルファベット順である
遺伝子特異的な実験室の技術に関する考慮事項.
RAD21遺伝子のエクソン14は分節重複内にある。
遺伝子ごとの注目すべきバリアント
.大多数が新生バリアントだとすると、これらのバリアントの箇所は一般的に個々の患者で異なる。しかし、まれにSMC1Aの反復性の病的バリアントが存在する;表9を参照。
表9.
コルネリア デ ランゲ症候群:注目すべきSMC1A 病的バリアント| 参照配列 | DNAヌクレオチドの変化 | 予測されるたんぱく質の変化 | 参照文献 |
|---|---|---|---|
| NM_006306.3 | c.1487G>A | p.Arg496His | Deardorff et al [2007]、Huisman et al [2017] |
| c.1904G>A | p.Arg635His | Huisman et al [2017] |
表に掲載されたバリアントは、著者によって提供されている。GeneReviewsのスタッフは、バリアントの分類を独自には検証していない。
GeneReviewsは、ヒトゲノムバリエーション学会(varnomen.hgvs.org)の標準命名会議に従う。命名法の説明については、クイックリファレンスを参照。
更新履歴:
- Gene Review著者: Matthew A Deardorff, MD, PhD, Dinah M Clark, MS, Ian D Krantz, MD
日本語訳者: 牧優子、升野光雄、山内泰子、黒木良和
(川崎医療福祉大学大学院医療福祉学研究科遺伝カウンセリングコース)
Gene Review 最終更新日: 2006.8.14. 日本語訳最終更新日: 2011.9.23. -
GeneReviews著者: Matthew A Deardorff, MD, phD, Sarah E Noon, MS, and lan D Krantz, MD
日本語訳者:疋田美那子、升野光雄、山内泰子、黒木良和(川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
GeneReviews最終更新日: 2020.10.15. 日本語訳最終更新日: 2021.12.2.[ in present]
![]()