鰓眼顔症候群
(Branchiooculofacial Syndrome)
[Synonyms:BOF Syndrome]
Gene Reviews著者: Angela E Lin, MD, FAAP, FACMG, Chad R Haldeman-Englert, MD, and Jeff M Milunsky, MD, FACMG.
日本語訳者:佐藤康守(たい矯正歯科)、水上都、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2023.9.28. 日本語訳最終更新日: 2024.4.20.
原文: Branchiooculofacial Syndrome
要約
疾患の特徴
鰓眼顔症候群(BOFS)は、鰓(頸部ないし耳介の上方ないし下方)の皮膚欠損(これは、ほとんどわからないくらいごくわずかに皮膚が薄くなっている状態ないしヘアパッチから、紅斑状の「血管腫様」病変、さらには大きな滲出性糜爛に至るまで、大きな幅をもつ)、小眼球・無眼球・コロボーマ・白内障・鼻涙管狭窄/閉鎖などの眼奇形、長頭症・眼間開離ないし内眼角開離・広い鼻尖・眼瞼裂斜上・時として口蓋裂を伴う口唇裂や口唇形成術後を思わせる目立つ人中稜(以前、これは偽性口唇裂と呼ばれていた)・上口唇瘻・下顔面の筋力低下(啼泣時の顔の非対称や顔面神経領域の筋力低下といった顔面奇形を特徴とする疾患である。
耳介奇形ないし目立つ耳介、内耳奇形や錐体骨奇形に起因する難聴も多くみられる。
知能はふつう正常である。
診断・検査
発端者におけるBOFSの診断は、特徴的な臨床所見を有することに加え、分子遺伝学的検査にてTFAP2Aのヘテロ接合性病的バリアントを同定することで確定する。
臨床的マネジメント
症状に対する治療:
BOFS罹患児の管理は、一般に、頭蓋顔面分野を専門とする医師、形成外科医、耳鼻咽喉科医、言語治療士などの多職種チームによって行っていく必要がある。皮膚欠損は、ごく小さな線状、表在性のものであれば、自然に治癒することもあるが、中には外科的介入を要するようなものもある。眼症候の治療は小児眼科医の手で行う。鼻涙管の狭窄や閉鎖については、手術が必要になることが多い。無眼球や小眼球の場合は、コンフォーマー(眼窩内に挿入して眼窩の成長を促す器材で、多くはプラスチック製)が必要になることがある。口唇裂については、経験豊富な小児形成外科医によって行うことが推奨される。鼻尖の異常、口唇裂の軽症型(いわゆる「偽性口唇裂」)、耳介奇形についても、手術が必要になることがある。難聴、腎奇形、歯の症候、先天性心疾患については標準治療を行う。感覚、心理、発達の各方面の課題については、支持療法を行う。
定期的追跡評価 :
- 眼科医の推奨に従って行う眼科的検査と視覚の評価
- 耳鼻咽喉科医ないし聴覚士の推奨に従って行う聴覚評価
- 膀胱尿管逆流を疑わせるような反復性尿路感染症の評価、歯のサイズ、数、齲蝕、咬合異常に関する評価、ならびに新たな嚢胞に関する評価を来院ごとに
- 発達と行動に関する評価を年に1度あるいは必要に応じて
- 思春期に入ろうとしている年長の小児については、自尊心の低下や心理学的問題の徴候のモニタリングを来院ごとに
遺伝カウンセリング
BOFSは、常染色体顕性の遺伝形式をとる。罹患者の50%-60%はde novoの病的バリアントに起因するものである。BOFS罹患者の子に病的バリアントが継承される確率は50%である。家系内に存在するTFAP2Aの病的バリアントが同定されている場合は、出生前検査や、着床前遺伝学的診断を行うことが可能である。
診断
鰓眼顔症候群(BOFS)については、現在のところ、コンセンサスパネルの作成した診断のための正式なガイドライン、臨床所見を階層化したアルゴリズム、エビデンスに基づいた検査基準といったものは存在しない。診断基準として提唱されたものが存在する(Milunskyら[2011]の表Ⅰを参照)。
本疾患を示唆する所見
次のカテゴリーの中の2つあるいは3つのカテゴリーに該当所見がみられる発端者については、鰓眼顔症候群(BOFS)を疑う必要がある。
鰓(皮膚)欠損
- 頸部ないし耳介下部、耳介上部の皮膚欠損
- ほとんどわからないくらいわずかな皮膚の菲薄化やヘアパッチから、紅斑性の「血管腫様」病変や大きな滲出性糜爛に至るまで、幅がみられる。
- 両側性で、かつ頸部前方部分にある場合が最もわかりやすく、「皮膚欠損症(cutis aplasia)」として報告されることもある[Wurfbainら2023]。
- 鰓耳腎症候群(BOR症候群)でみられるような刺し傷様の瘻管とは明確に異なる。
- ごく軽度の例では、発見しづらく、また自然治癒することもあるが、多くは滲出性になっていく傾向がある。
眼の奇形
- 小眼球,無眼球
- コロボーマ
- 白内障
- 眼瞼下垂
- 鼻涙管狭窄/閉鎖
- 斜視
顔の異常
- 長頭症、眼間開離ないし内眼角開離、広い鼻尖、眼瞼裂斜上を伴う特徴的顔貌(図1)
- 時に口蓋裂を伴うこともある口唇裂ないし目立つ人中稜(学術的には口唇裂の軽度型で、以前は「偽性口唇裂」と呼ばれていた)(口蓋裂単独の例は報告されていない)
- 上口唇瘻
- 下部顔面神経ないしその部の筋の低形成(啼泣時の顔の非対称、顔面神経領域の筋力低下)
- 蝸牛異形成、Mondini型奇形、前庭水管拡大などの内耳・錐体骨奇形
- 耳介奇形ないし目立つ耳介
- 難聴(伝音性、感音性、混合性)
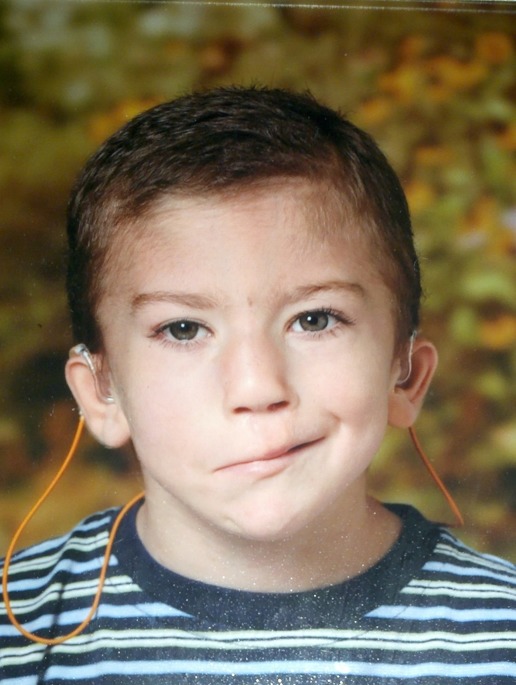
図1:BOF症候群5歳男児の写真
分子レベルの検査結果の詳細については、Milunskyら[2008](症例3:2歳)で報告されている。本症例では、すでに修復術施行済の右側頸部皮膚欠損(B)、両側性の鼻涙管狭窄と眼間開離(O)、両側性の「痕跡」唇裂と広い鼻尖、軽度の眼瞼裂斜上(F)を呈する。両側性の骨導性難聴があるものの、患者本人が骨導補聴器ではなく従来型の補聴器を選択した。スマイルにて、右側の下部顔面神経領域の筋力低下があることがわかる。幼稚園に通っているが、聴覚障害者学習センターの個別教育計画の目標をすべて達成している。非常に社交的で、英語のスピーチやアメリカ手話の習得も良好である。
この写真は、母親のご厚意により提供されたものである。
診断の確定
発端者におけるBOFSの臨床診断は、提唱されている臨床診断基準[Milunskyら2011]を満たすことで確定し、発端者におけるBOFSの分子診断は、分子遺伝学的検査でTFAP2Aにヘテロ接合性の病的バリアント(pathogenicとlikely pathogenicの両方を含む)が同定されることで確定する(表1参照)。
注:(1)アメリカ臨床遺伝ゲノム学会(ACMG)/分子病理学会(AMP)のバリアントの解釈に関するガイドラインによると、「pathogenic」のバリアントと「likely pathogenic」のバリアントとは臨床の場では同義であり、ともに診断に供しうるものであると同時に、臨床的な意思決定に使用しうるものとされている[Richardsら2015]。本セクションで「病的バリアント」と言うとき、それは、あらゆるlikely pathogenicのバリアントまでを包含するものと理解されたい。
(2)TFAP2Aにヘテロ接合性の意義不明バリアントが同定された場合、それは、本疾患の診断を確定するものでも否定するものでもない。
臨床診断
次の3つの主要症候がすべて存在すること
- 鰓の皮膚欠損
- 眼奇形
- 顔面奇形(特徴的顔貌)
もしくは、主要症候3つのうちの2つに加えて、以下のものの1つが存在すること
- 別個に診断が下された第1度近親が存在すること
- 異所性胸腺(皮膚)
分子診断
分子遺伝学的検査のアプローチとしては、遺伝子標的型検査(単一遺伝子検査、マルチ遺伝子パネル)、網羅的ゲノム検査(エクソームシーケンシング、ゲノムシーケンシング)を組み合わせるやり方が考えられる。遺伝子標的型検査は、臨床医が原因遺伝子の目星をつけた上で進める必要がある(「方法1」参照)が、網羅的ゲノム検査の場合、その必要はない(「方法2」参照)。
方法1
- 単一遺伝子検査
ミスセンス・ナンセンス・スプライス部位の各バリアントや、遺伝子内小欠失/挿入の検出を目的として、最初にTFAP2Aの配列解析を行う。
注:用いる配列解析の手法によっては、単一エクソン、複数エクソン、遺伝子全体といった単位の欠失/重複が検出されないことがある。配列解析を行ってもバリアントが検出されないときは、次のステップとして、エクソン単位あるいは遺伝子全体の欠失/重複を検出するための遺伝子標的型欠失/重複解析を行うようにする。
- マルチ遺伝子パネル検査
現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝学的原因を特定することを目的として、TFAP2Aその他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルも検討対象になろう。
注:(1)パネルに含められる遺伝子の種類、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
方法2
- 網羅的ゲノム検査の場合は、臨床医の側で事前に原因遺伝子の目星をつけておく必要はない。エクソームシーケンシングが最も広く用いられているが、ゲノムシーケンシングを用いることも可能である。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:鰓眼顔症候群で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的バリアント2が検出される発端者の割合 |
|---|---|---|
| TFAP2A | 配列解析3 | 95%超4 |
| 遺伝子標的型欠失/重複解析5 | 5%未満6 |
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子において検出されるバリアントの情報については、「分子遺伝学」の項を参照。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失や重複については検出されない。配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- Milunskyら[2011]
- 遺伝子標的型の欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失ないし重複の検出を目的に設計された遺伝子標的型マイクロアレイなどがある。
- Milunskyら[2008],Gestriら[2009]
臨床的特徴
臨床像
鰓眼顔症候群罹患者の大多数は、臨床症候をもとに乳児期に診断を行うことが可能である。罹患者の男女比は1:1である。顔の特徴は認識可能なレベルであることが多いものの、症例によっては微妙な違いにとどまるようなこともある[著者の個人的観察]。
鰓(皮膚)欠損
欠損のみられる部位は、頸部(90%)、あるいは耳介の下部ないし上部(60%)である。
- 欠損には、ほとんどわからないくらいごくわずかに皮膚が薄くなっている状態ないしヘアパッチから、紅斑状の「血管腫様」病変、さらには大きな滲出性糜爛に至るまで、大きな幅がみられる。
- ごく軽微な欠損の場合は気づかれることなく経過し、稀には完全に自然治癒してしまうことがある。時に小さな腔や管が残っていることがあり、そこから「流涙」様の浸出液が出ることで穴の開いていることが明らかになるようなことがある。
眼の奇形
眼の奇形としては次のようなものがある。
- 眼の構造的奇形としては、小眼球、無眼球、コロボーマ、白内障などがある。
- 眼周囲の異常としては、涙目をきたすような鼻涙管狭窄/閉鎖や眼瞼下垂などがある。
- 視覚関連の問題としては、斜視や重度の視覚障害などがある。
- Lamら[2023]は、BOFSとしてそれまでに報告されていた172例に新たに1例を加えて、その眼所見に関するレビューを行っている。
そしてその結果、最も多くみられた所見は、鼻涙管狭窄(57%)、コロボーマ(46%)、無眼球/小眼球(37%)、白内障(16%)、斜視(14%)、近視(12%)であったとしている。
顔の奇形
特徴的症候としては、長頭症、眼間開離ないし内眼角開離、広い鼻尖、眼瞼裂斜上がある(図1参照)。その他の所見としては、次のようなものがある。
- 口唇裂ないし目立つ人中稜(学術的には痕跡唇裂の1型として知られ、以前は「偽性口唇裂」と呼ばれた)
- これに口蓋裂が伴うこともあれば伴わないこともある(99%)
- 口唇裂のない口蓋裂単独例は報告されていない。
- 上口唇瘻
- 下顔面の顔面神経/筋の低形成(啼泣時の顔の非対称、顔面神経領域の部分的筋力低下)
- 耳の奇形
- 耳介奇形ないし目立つ耳介
- 蝸牛異形成、Mondini型奇形、前庭水管拡大などの内耳・錐体骨奇形
- 難聴(70%)(伝音性、感音性、混合性)
- 通常の口唇裂患者にみられるものとははっきり異なる広い鼻尖を伴う幅広の鼻
BOF症候群でみられるその他の所見
免疫系
胸腺奇形(異所性、皮膚)(約35%)が通常、両側性に現れるが、胸腺機能は正常。
泌尿器系
- 構造奇形(35%)(異形成腎、腎無形成、多嚢胞性腎など)
- 膀胱尿管逆流
外胚葉(毛髪,歯,爪)
- 早発性白髪,白毛症(forelockもしくはパッチ状)(35%)
- 歯の低形成
- 爪の異形成
- 皮下の嚢胞(類皮腫様で、しばしば頭皮上に出現;他の頭頸部への出現は比較的少ない)
精神運動発達(通常は正常)
- 視覚や聴覚の障害(頻発)
- 自閉症スペクトラム障害,知的障害(稀)
成長障害
それほど多くはない。
その他、雑多で稀な(報告数5例未満)問題
- 虹彩異色症
- 先天性心疾患(心房中隔欠損,Fallot四徴)
- 多指趾症(両側性で、通常は軸後性)
- 髄芽腫(1例)[Milunskyら2008]
- 三角頭蓋(1例)[Wurfbainら2023]
遺伝型-表現型相関
明確な遺伝型-表現型相関はみられない。
同一の病的バリアントであっても、家系間、家系内で症候の現れ方には大きな幅がみられる[Milunskyら2011]。一方、ミスセンス、フレームシフト、スプライス部位の各バリアント、遺伝子全体のより複雑な再構成[Tekinら2009,Milunskyら2011]などのいずれもが、同じような表現型となって現れる。
ただ、TFAP2A領域の関与する欠失を有する罹患者については、その大多数が口唇裂の軽微型のスペクトラム上にあると思われる目立つ人中稜を呈するようである[Linら2009]。Le Blancら[2013]は、TFAP2Aを含む計6つの遺伝子にまたがる593kbの欠失を有する母と乳児の例を報告している。その母子については、2人とも口唇裂や人中の異常はみられなかったと報告されている。この点を除けば、遺伝子内に生じた病的バリアントに起因して現れる表現型に家系間、家系内で大きな幅がみられるのと全く同じように、TFAP2Aの欠失の場合も、やはり表現型には大きなばらつきが存在するようである。
浸透率
BOFSの浸透率は、ほぼ100%であることがわかっている。TFAP2Aの病的バリアントを有することが確認されたBOFSの家系に属する人たちについては、早発性白髪(毛染めで隠している可能性がある)、腰のない頸部の毛、虹彩異色といったわずかな症候がないかどうか、慎重に診査する必要がある。
頻度
BOFSの発生頻度は明らかではない。BOFSは稀な疾患で、臨床診断や分子診断が詳細に報告されたものは150例に満たない。2017年の顔面形態異常学会に出席した臨床遺伝医を対象に非公式な調査を行ったところ、27人の未発表症例(臨床診断が18例、分子診断が9例)があることが判明した。人口ベースでの発生頻度を算出する上では十分な数ではなく、BOFSが依然、稀な疾患であるという印象を追認するものとなっている。
遺伝学的に関連のある疾患(同一アレル疾患)
TFAP2Aの病的バリアントに関連するものとしては、このGeneReiewで述べたもの以外の表現型は知られていない。
鑑別診断
鰓眼顔症候群の表現型は独特であるため、一部の症候が重なる他の疾患とは、多くの場合、臨床の場で鑑別可能である(表2参照)。
表2:鰓眼顔症候群との鑑別を検討すべき疾患
| 遺伝子/遺伝学的メカニズム | 疾患名 | 遺伝形式 | その疾患でみられる症候 | |
|---|---|---|---|---|
| BOFSと重なる症候 | BOFSと異なる症候 | |||
| 22q11.2欠失 | 22q11.2欠失症候群 | AD |
|
|
| CHD7 | CHD7疾患(CHARGE症候群を含む) | AD |
|
|
| EDN3 EDNRB KITLG MITF PAX3 SNAI2 SOX10 |
Waardenburg症候群(「Waardenburg症候群Ⅰ型」のGeneReviewを参照) | AD AR |
|
|
| EYA1 SIX5 SIX1 |
鰓耳腎スペクトラム障害 | AD |
|
|
| TP63 | EEC症候群3型(「TP63関連疾患」のGeneReviewを参照) | AD |
|
|
AD=常染色体顕性;AR=常染色体潜性
臨床的マネジメント
最初の診断に続いて行う評価
鰓眼顔症候群(BOFS)と診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済でないようなら、表3にまとめた評価を行うことが推奨される。
表3:鰓眼顔症候群:最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 |
コメント |
|---|---|---|
鰓欠損 |
小児形成外科医による皮膚欠損の診査 |
病変の範囲の確定、洞の有無の確認に加え、最も重要なのは胸腺組織の遺残の確認。 |
眼 |
小児眼科医による眼の詳細な検査 |
視覚障害、斜視、鼻涙管閉塞に関する評価。 |
無眼球や重度の小眼球を有する例について、視覚障害者支援サービスへの紹介 |
|
|
口唇裂口蓋裂 |
口唇口蓋裂その他の顔面の異常に関する正式な評価 |
口唇口蓋裂チームの手で行う。 |
聴覚 |
|
|
腎 |
腎異常が確認された場合は、腎臓専門医への紹介とともに腎超音波検査 |
|
歯 |
歯に関するあらゆることの評価を目的とした紹介 |
裂の評価の一環として、すでに行われている場合を除く。 |
神経発達/行動 |
|
|
心臓 |
心雑音や心症状がある例については心エコー |
|
遺伝カウンセリング |
遺伝の専門医療職2が行う。 |
医学的、個人的な意思決定の用に資するべく、本人や家族に対し、BOFSの本質、遺伝形式、そのもつ意味についての情報提供を行う。 |
家族への支援/情報資源 |
以下の必要性に関する評価
|
|
- Ravehら[2000],Stoetzelら[2009],Tekinら[2009]
- 臨床遺伝医、認定遺伝カウンセラー、認定上級遺伝看護師をいう。
注:(1)BOFSでは運動機能の遅れはみられない。したがって、理学療法や作業療法は不要である。
(2)がんの追跡評価を行う意味については、現時点ではよくわかっていない。
症候に対する治療
Milunskyら[2011]による管理のガイドラインがある。これは現在でも有用なものではあるが、アップデートはなされていない。一般に、BOFSと多発奇形を有する子どもは、多職種によるケアが提供可能な環境でフォローがなされるべきである。そのチームの構成は、例えば、頭蓋顔面分野を専門とする医師、形成外科医、耳鼻咽喉科医、言語治療士といったものである(表4参照)。理想を言えば、そうした多職種による評価や手術は、1つの頭蓋顔面医療機関の中で行われるのが望ましい。
表4:鰓眼顔症候群:症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 鰓欠損 |
|
鰓部あるいは耳介上部の皮膚欠損で、小さく、線状、表在性のものについては、自然治癒の可能性あり。 |
| 瘻管は、熟練した小児形成外科医の手で切除する必要あり。 |
|
|
| 眼症候 | 小児眼科医による管理 |
|
| 口唇裂口蓋裂 |
|
口唇裂の軽症型(以前、「偽性口唇裂」と呼ばれていたもの)についても修正術が必要となる場合あり1。 |
| 耳/聴覚 |
|
乳児期初期に診断がなされた場合には、耳介矯正が適用できる場合あり。 |
| 腎症候 | 腎臓専門医/泌尿器科医による標準治療 | |
| 歯の症候 | 歯科医/矯正歯科医による標準治療 | |
| 神経発達/行動 | 感覚、心理、発達上の問題に対しては支持療法での対応が必要。 | 重症者向けの心理サポートがもっと必要かどうかという点に関しては、裏づけとなるデータが不足している現状である。 |
| 先天性心疾患 | 心臓病専門医による標準治療 |
Milunskyら[2011]の表Ⅳより引用。
- Linら[2009]
定期的追跡評価
現段階でみられる症候の様相、ならびに支持療法に対する反応様相のモニタリングを目的として、表5に示した評価が推奨される。
表5:鰓眼顔症候群:推奨される定期的追跡評価
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 眼/視覚 | 眼科的検査と視覚評価 | 眼科医に推奨に従って。 |
| 聴覚 | 聴覚評価 | 耳鼻咽喉科医/聴覚士の推奨に従って。 |
| 腎症候 | 膀胱尿管逆流を示唆する反復性尿路感染症の評価 | 来院ごと。 |
| 歯 | 歯のサイズ、数、齲蝕、咬合異常に関するモニタリング | |
| 皮膚 | 新たな嚢胞に関する評価 | |
| 神経発達/行動/精神 | 発達と行動の評価 | 年に1度あるいは必要に応じて。 |
| 自尊心の低下その他の心理的問題の徴候に関するモニタリング | 思春期に入りつつある年長の小児について来院ごと。 |
リスクを有する血縁者の評価
聴覚、視覚、腎その他の症候に関して、定期的追跡や積極的治療を行うことで便益が得られる人を可能な限り早期に特定することを目的として、リスクを有する血族で、見かけ上、無症候の人たちについては、罹患者より年上であるか年下であるかを問わず、評価を行うことが望ましい。
評価の内容としては、以下が考えられる。
- 家系内に存在する病的バリアントが既知の場合は、分子遺伝学的検査
- 家系内における病的バリアントが同定されていない場合は、BOFSをうかがわせるごくわずかな身体的所見がないかどうかを慎重にチェックするための診査
遺伝カウンセリングを目的として、リスクを有する血族に対して行う検査関連のことについては、「遺伝カウンセリング」の項を参照のこと。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、米国の「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
鰓眼顔症候群(BOFS)は常染色体顕性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
- BOFSの診断の下りた罹患者の40-50%については、その片親が罹患者である[Milunskyら2011]。
- BOFSの診断の下りた罹患者の50-60%については、TFAP2Aに生じたde novoの病的バリアントに起因するものである[Milunskyら2011]。
- 分子診断が確定し、かつ家系内で唯一の罹患例(すなわち、孤発例)と目される発端者については、発端者の両親に対しても遺伝学的検査を行い、その遺伝学的状態を確認するとともに再発リスクに関するカウンセリングの信頼性を確保するようにすることが推奨される。
- 発端者で同定された病的バリアントが、両親いずれの白血球DNAからも検出されず、なおかつ、親子鑑定の結果、両者が真正の生物学的父・母であることが確認されている場合は、以下のようなことが可能性として考えられる。
- 発端者に生じたde novoの病的バリアントの可能性。
- 生殖細胞系列モザイク(あるいは、体細胞・生殖細胞系列両方のモザイク)の片親から発端者が病的バリアントを継承した可能性*。
注:親の白血球DNAを検査しても、体細胞モザイクの全例を検出できるわけではない。
また、生殖細胞のみに存在する病的バリアントについては、一切検出されない。
*TFAP2Aの病的バリアントを体細胞・生殖細胞系列両方のモザイクで有する片親については、軽度ないしごく軽微な罹患にとどまっていることが考えられる[Milunskyら2011]。
- BOFSと診断された罹患者の家族歴については、家系内の人が本疾患を有していることを見逃してしまった、あるいは、家系内の人の症候がごく軽微なものであったといった理由で見かけ上、陰性のように思われることがある。したがって、見かけ上、陰性のように思われても、分子遺伝学的検査にて、両親とも、発端者で同定された病的バリアントのヘテロ接合体ではないことを確認しない限り、家族歴陰性の確定はできない。
発端者の同胞
発端者の同胞の有するリスクは、発端者の両親の遺伝学的状況によって変わってくる。
- 発端者の片親が罹患者であった、ないしは、発端者で同定された病的バリアントを片親も有していたといった場合、同胞の有するリスクは50%である。BOFSの浸透率はほぼ100%であるが、家系内における表現型のばらつきはかなり大きいことがわかっている[Milunskyら2011]。
- 発端者で同定されたTFAP2Aの病的バリアントが、両親いずれの白血球DNAからも検出されない場合、同胞への再発リスクは一般集団より若干高い程度と推定される。それは、親の生殖細胞系列モザイクの可能性が考えられるからである[Milunskyら2011]。
- 両親とも、臨床的にみて非罹患者と目されるものの、その遺伝学的状況まではわかっていないという場合、発端者の同胞の有するリスクは低いと思われるものの、一般集団の値よりは高くなる。それは、実際はヘテロ接合を有している片親の表現型が軽度にとどまった可能性、ならびに、片親の生殖細胞系列モザイクの可能性が残るからである。
発端者の子
BOFS罹患者の子がTFAP2Aの病的バリアントを継承する可能性は50%である。
他の家族構成員
他の血族の有するリスクは、発端者の母親の状況によって変わってくる。仮に母親もOFD1の病的バリアントを有していた他の血族の有するリスクは、発端者の両親の遺伝子の状態によって変わってくる。片親がTFAP2Aの病的バリアントを有していた場合、その片親の血族にあたる人はすべてリスクを有することになる。
関連する遺伝カウンセリング上の諸事項
早期診断・早期治療を目的として、リスクを有する血族に対して行う評価関連の情報については、「臨床的マネジメント」の中の「リスクを有する血縁者の評価」の項を参照されたい。
家族計画
- 遺伝的リスクの確定、ならびに出生前検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- 若い成人の罹患者に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原メカニズム・疾患等に対するわれわれの理解は、将来、より進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった病原メカニズムが未解明の)発端者のDNAについては、保存しておくことを検討すべきである。
詳しくは、Huangら[2022]を参照されたい。
出生前検査ならびに着床前遺伝学的検査
家系内に存在するTFAP2Aの病的バリアントが判明している場合は、出生前検査ならびに着床前遺伝学的検査が可能である。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- American Cleft Palate-Craniofacial Association
Phone: 919-933-9044
www.acpa-cpf.org
- Face Equality International
United Kingdom
Email: info@faceequalityinternational.org
aceequalityinternational.org
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:鰓眼顔症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| TFAP2A | 6p24.3 | 転写因子AP-2-α | TFAP2A database | TFAP2A | TFAP2A |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:鰓眼顔症候群関連のOMIMエントリー内容の閲覧はOMIMへ)
| 107580 | TRANSCRIPTION FACTOR AP2-ALPHA; TFAP2A |
| 113620 | BRANCHIOOCULOFACIAL SYNDROME; BOFS |
分子レベルの病原
TFAP2Aは、転写因子AP-2ファミリーの1つの、レチノイン酸応答性タンパク質で、胚発生期に、眼、耳、顔、体壁、四肢、神経管において遺伝子発現の調節を行っている[Schorleら1996,Zhangら1996,Ahituvら2004,Nelson & Williams 2004]。TFAP2Aはまた、タンパク質発現レベルが細胞の形質転換、腫瘍の増殖、転移、生存に影響を及ぼす形で、腫瘍発生にも関与している[Jeanら1998,Heimbergerら2005,Orsoら2007]。TFAP2Aの関与する分子レベルの障害に起因して、さまざまな表現型が現れる背景には、おそらく数多くの遺伝子との相互作用が関わっているものと思われる。TFAP2Aは、遊走前ならびに遊走中の神経堤細胞において発現することが知られており[Hilger-Eversheimら2000,Li & Cornell 2007]、水晶体の形態形成初期に必要なものである[Gestriら2009]。
病的バリアントは、遺伝子全体に分布するものの、エクソン6と7に1つのホットスポットがあり、この領域のミスセンスバリアントが、BOFSの発端者/家系の約90%を占めることが明らかになっている[Milunskyら2011]。
Liら[2013]は、DNA結合ドメインの病的バリアントのいくつかについて、野生型AP-2αタンパク質に対してドミナントネガティブ効果をもつ可能性があることを明らかにしている。こうしたことから、ヌルアレル(訳注:野生型の本来有している機能を全く有しない産物を生成するアレル)、ハイポモルフィックアレル(訳注:野生型の本来有している機能が低下した産物を生成するアレル)、アンチモルフィックアレル(訳注:野生型の本来有している機能を阻害するような産物を生成するアレル)といった活性の違いが、BOFSの表現型のばらつきの違いとなって現れている可能性が考えられる。
疾患の発症メカニズム
機能喪失型である。
更新履歴:
-
>GeneReviews著者: Angela E Lin, MD, FAAP, FACMG,Chad R Haldeman-Englert, MD,Jeff M Milunsky, MD, FACMG
日本語訳者:佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日:2018.3.29. 日本語訳最終更新日: 2022.6.29. - Gene Reviews著者: Angela E Lin, MD, FAAP, FACMG, Chad R Haldeman-Englert, MD, and Jeff M Milunsky, MD, FACMG.
日本語訳者:佐藤康守(たい矯正歯科)、水上都、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2023.9.28. 日本語訳最終更新日: 2024.4.20.[in present]
![]()