Beckwith-Wiedemann症候群
(Beckwith-Wiedemann Syndrome)
[Synonyms: Wiedemann-Beckwith症候群]]
Gene Reviews著者: Cheryl Shuman, MS, CGC, J Bruce Beckwith, MD, and Rosanna Weksberg, MD, PhD, FRCPC, FCCMG, FACMG
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2016.8.11. 日本語訳最終更新日: 2022.8.1.
原文 Beckwith-Wiedemann Syndrome
要約
疾患の特徴
Beckwith-Wiedemann症候群(BWS)は、新生児低血糖、巨軀、巨舌、半側過形成、臍帯ヘルニア、胎児性腫瘍(例えば、Wilms腫瘍,肝芽腫,神経芽腫,横紋筋肉腫)、内臓巨大症、副腎皮質細胞腫大、腎異常(例えば、髄質異形成,腎石灰化症,髄質海綿腎,腎肥大)、外耳の皺/瘻孔などがさまざまな幅で現れる成長障害である。
BWSは一種の臨床的スペクトラムと考えるべきもので、上記の症候の多くを有する罹患者がいる一方で、1つ、2つの臨床症候しか示さない例もみられる。
早産、低血糖、心筋症、巨舌、腫瘍等の合併症により、早期死亡に至ることもある。
ただ、以前に報告されていた20%という死亡率については、この疾患の理解が進み、治療上の選択肢も増えた今となっては、過大な数字であるように思われる。
巨舌と巨軀は、通常、出生時にみられるものの、出生後に始まる例もみられる。
成長速度は7-8歳には鈍化する。
半側過形成は、身体の一部分に分節状に現れることもあれば、特定の器官・組織に現れるようなこともある。
診断・検査
臨床評価に基づいて下されたBWSの予備的診断を、分子遺伝学的/細胞遺伝学的検査で確認するといった形が考えられる。
細胞遺伝学的に検出可能な形での11p15の異常を有する例は、罹患者全体の1%以下である。
一方、11p15領域のエピゲノムやゲノムの以下のような変化に起因して生じたBWSについては、分子遺伝学的検査により同定が可能である。
- 罹患者の50%にみられる母性染色体のインプリンティングセンター2(IC2)の低メチル化。
- 罹患者の20%にみられる11p15領域の父性片親性ダイソミー。
- 罹患者の5%にみられる母性染色体のインプリンティングセンター1(IC1)のメチル化。
この領域の欠失ないし重複に伴ってメチル化の変化が生じている場合は、高い継承性を示す。
CDKN1Cの配列解析を行うことで、家族性BWSの約40%、非家族性BWSの5%-10%に、母性継承性の病的バリアントのヘテロ接合が検出される。
臨床的マネジメント
症状に対する治療:
中枢神経系の合併症リスクを低減するための低血糖の治療、臍帯ヘルニアに対する腹壁修復、気道障害に対応するための気管内挿管、巨舌に起因する摂食障害に対応するための特殊乳首や経鼻胃管栄養の使用などがある。
巨舌を有する子どもに対しては、場合によっては、乳幼児期に舌縮小術とともに言語治療を行うことが有益である。
半側過形成に伴って二次性に生じる下肢長の左右差に対しては、思春期初期に均等化手術が行われることがある。
また、顔面の半側過形成を呈する罹患者に対しては、頭蓋顔面形成術が有益になることもあろう。
新生物に関しては、小児腫瘍学の標準プロトコルに従って治療が行われる。
腎石灰化症その他の腎症候に対しては、小児腎臓内科医の手で評価や治療を行う必要がある。
消化管の器質的奇形を有する子どもについては、関連専門分野への紹介を行う。
心臓の問題に関しては、標準治療を行う。
発達遅滞を有する子どもに対しては、標準介入を行う。
二次的な合併症の予防:
尿路感染症が疑われる場合には、二次性の腎障害を予防する観点から、評価ならびに標準治療を迅速に行う。
定期的追跡評価 :
特に新生児期においては、低血糖に関するモニタリングを行う。
腹部超音波検査による胎児性腫瘍のスクリーニングを、3ヵ月ごとに8歳まで行う。
肝芽腫の早期発見を目的として、満4歳に達するまでは、2-3ヵ月ごとに血清αフェトプロテイン(AFP)濃度のモニタリングを行う。
8歳から思春期中期までの罹患者については、腎石灰化症や髄質海綿腎を有する罹患者を同定するため、腎超音波検査を年に1度行う。
カルシウム/クレアチニン比の測定を年に1、2度行うことを検討する。
遺伝カウンセリング
Beckwith-Wiedemann症候群は、11p15.5領域にある2つのインプリントドメインにおける遺伝子の転写制御の異常によって生じる疾患である。
BWS罹患者の大多数は、染色体検査、すなわち核型の点では正常である。
BWS罹患者の約85%はBWSの家族歴を有しない一方、約15%は、常染色体顕性遺伝に一致した形での親由来の伝達を示す家族歴を有する。
親の低妊孕性のため生殖補助医療(ART)によって誕生した子どもについては、BWSを含むインプリンティング障害のリスクが高まる可能性がある。
再発リスクの推定に関しては、背景にあるBWSの原因となった遺伝メカニズムを解明することで、より向上させることができる。
一般妊婦を対象とした出生前スクリーニングでBWSを示唆する所見がみられた場合は、染色体分析、染色体マイクロアレイ、分子遺伝学的検査といったものも検討の対象となろう。
染色体異常を継承している家系については染色体分析を、BWSの分子メカニズムが解明済の家系については分子遺伝学的検査をといった形で、特異的な出生前検査を行うことが可能である。
診断
本疾患を示唆する所見
Beckwith-Wiedemann症候群(BWS)の臨床的診断基準に関する合意は得られていない。
以下に示す大症候、小症候を1つ以上有する例については、Beckwith-Wiedemann症候群を疑う必要がある。
BWSで現れる大症候
- 巨軀(以前より、97パーセンタイル超の体重・身長が基準とされている)
- 巨舌
- 半側過形成(身体の1領域ないし複数領域に生じる非対称性の過成長)
- 臍帯ヘルニアまたは臍ヘルニア
- 子どもにみられる胎児性腫瘍(例えば、Wilms腫瘍,肝芽腫,神経芽腫,横紋筋肉腫)
- 肝・脾・腎・副腎・膵等の腹腔内臓器の1つないし複数に生じる内臓巨大症
- 胎児性副腎皮質細胞腫大(これは本疾患に特異的な症候である)
- 腎の器質的異常、腎肥大、腎石灰化症や、後に髄質海綿腎に移行していくものを含めた腎の異常
- 耳垂前部の直線状の皺、ないし後部耳輪の瘻孔
- 間葉性異形成胎盤[Wilsonら2008]
- 口蓋裂(BWSでは稀)
- 心筋症(BWSでは稀)
- 家族歴陽性(BWSの臨床診断を受けた例、あるいは、BWSを示唆する病歴や症候を有する血族が1人以上いること)
BWSで現れる小症候
- 羊水過多、早産等の妊娠関連所見
- 新生児低血糖
- 単純性母斑(通常、前額部、眉間、後頸部に現れる)あるいは血管腫(皮膚性・皮膚外性)といった血管病変
- 中顔面の後退や眼窩下部の溝といった特徴的顔貌
- 構造的心奇形あるいは心拡大
- 腹直筋離開
- 骨年齢の亢進(過成長/内分泌障害で多くみられる)
診断の確定
発端者におけるBWSの診断は、以下のいずれかで確定する。
- 大症候3つ、あるいは、大症候2つに加えて1つ以上の小症候がみられること(「本疾患を示唆する所見」の項を参照)
注:BWSは1つの臨床的スペクトラムとみなすべきものであり、罹患者によっては、本疾患を示唆する臨床所見を1つか2つしか有しないようなこともある。
したがって、一般に受け入れられている臨床的基準としてここで紹介したものは、絶対的基準とみなすべきものではなく、むしろガイドラインと捉えるべきものである。
言い換えれば、こうしたものはBWSの診断を否定する目的で使用することはできず、また、臨床的判断の代用になりうる性質のものでもない。
- 1つ以上の臨床症候がみられることに加え、11p15.5のメチル化異常をもたらすエピゲノムないしゲノムの変化、もしくはBWSを引き起こすCDKN1Cの病的バリアントのヘテロ接合が確認されること(以下の記述ならびに表1を参照)。
BWSは、11p15.5にある2つのインプリンティングを受けたドメイン(BWSクリティカル領域として知られる)における遺伝子転写制御の異常に伴って生じる。
こうした制御異常は、数多くあるメカニズムの中のいずれもがその原因となりうるものである。
ここでは、「遺伝学的検査」の項に記載した検査への道順を明確にすることを目的として、これまでに知られている病因のメカニズムをごく簡略に説明する。
この領域における遺伝子発現の制御に関する詳細については、「分子レベルの病原」の項を参照されたい。
BWSクリティカル領域内には2つのドメインが存在する。
ドメイン1に位置するIGF2やH19の発現は、インプリンティングセンター1(IC1)による制御を受ける。
一方、ドメイン2に位置するCDKN1C、KCNQ1OT1、KCNQ1の発現制御のほうはインプリンティングセンター2のほうが担っている(図1)。
ゲノムインプリンティングとは、1つの遺伝子内にある2つのアレルのDNAがそれぞれ異なった形で修飾を受けることで、遺伝子の中の片親由来のアレルだけが通常、発現するようになる現象をいう[Barlow1994]。
図1aに示したように、IC1とIC2が片親特異的にメチル化を受けることで、非罹患者においては、父性アレル・母性アレル上の特定の遺伝子のみが発現することとなる。
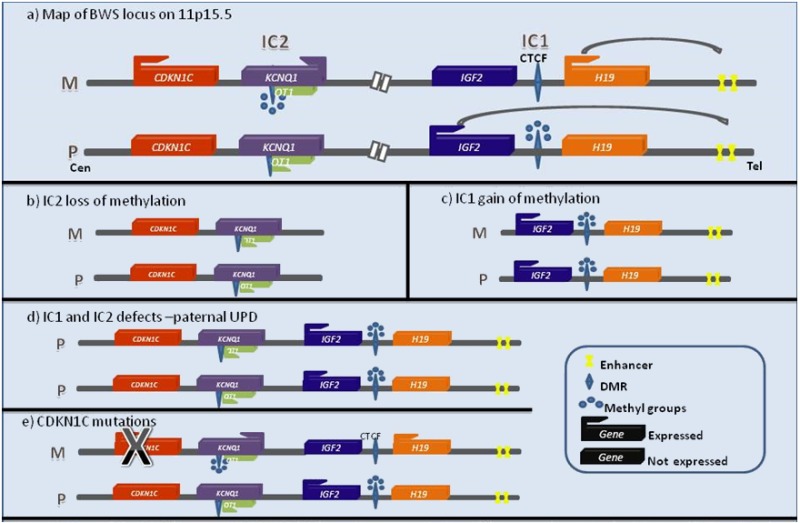
図1:11p15.5上に座位するBWSクリティカル領域を説明するマップ
a)は、正常な形で親起源特異的なインプリンティングがなされたアレルの発現様相を模式的に表したものである。
注:b)とc)は、変化が生じた部位だけを描いたものである。
IC=インプリンティングセンター,Cen=セントロメア,Tel=テロメア,P=父性,M=母性、
DMR=メチル化可変領域
OT1は、KCNQ1アンチセンス転写産物であるKCNQ1OT1を指す。
この図は、実際の距離に合わせた縮尺で描いたものではない。
(Choufaniら[2010]より、JohnWileyandSons社の許可を得て転載。)
注:IC1、IC2はそれぞれ、メチル化可変領域DMR1、DMR2と呼ばれることがある。
遺伝子検査を行うことで、BWS罹患者の80%超で、次の5つの変化のうちの1つが検出される[Weksbergら2003,Weksbergら2005]。
- 図1は、次の4種の分子的変化を模式的に示したものである。
- 母由来染色体のIC2の脱メチル化(図1b)
- 母由来染色体のIC1のメチル化(図1c)
- 11p15.5の父性片親性ダイソミー(図1d)
- 母由来CDKN1Cアレルに生じた病的バリアントのヘテロ接合(図1e)
- 次の遺伝子変化は、図1には示していない。
細胞遺伝学的に見える形での重複・逆位・転座等、11p15.5領域を含むゲノムバリアント、あるいは11p15.5の微小重複・微小欠失等のコピー数変化
注:上記のうち、母由来CDKN1Cアレルの病的バリアント以外のものについては、いずれも、ゲノムバリアントにメチル化状態の変化が関与していると思われるものである[Niemitzら2004,Sparagoら2004,Prawittら2005,Baskinら2014]。
遺伝的メカニズム別にみたBWS罹患者の割合を図2にまとめてみた。
図2:遺伝的メカニズム別にみたBeckwith-Wiedemann症候群の原因
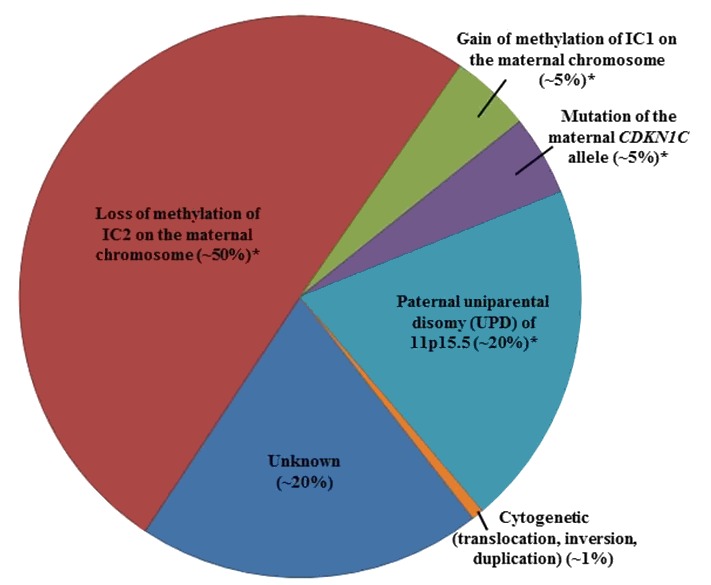
*分子的に分類したときのこれらのサブグループは、いずれもDNAメチル化の異常であるが、同時にまた、根底にあるゲノム変化の結果であるとも言えるものである。
そうしたゲノム異常は、IC1の高メチル化で最も多くみられ、IC2の低メチル化で最も少ない。
11p15.5にあるBWSクリティカル領域に限ったゲノム異常であれば、MS-MLPA法、あるいはその他さまざまなシーケンシングの手法で検出可能である。
欠失/重複の一部はCMAで検出されよう。
遺伝学的検査
伝学的検査としては、DNAメチル化検査、単一遺伝子検査、11p15.5領域の配列に関するコピー数解析、染色体マイクロアレイ、核型分析、BWSクリティカル領域を含むマルチ遺伝子パネルの使用などが考えられる。
- DNAメチル化検査
IC1とIC2のDNAメチル化検査は、同時に施行するようにする。
- IC1とIC2の両方にメチル化異常がある場合は、片親性ダイソミーが示唆される。
- 再発リスクを調べることが目的であれば、追加の遺伝学的検査を行って、メチル化異常の原因となったメカニズムを調べることも可能である(「遺伝カウンセリング」の項を参照)。
- 単一遺伝子検査
家族性の例、正中列の奇形(口蓋裂,後頭蓋窩の異常,臍ヘルニア,尿道下裂[Gardinerら2012,Brioudeら2015])を伴うBWSの例、臨床的にみてBWSの疑いがきわめて濃いものの11p15.5領域の細胞遺伝学的異常、コピー数変化、メチル化異常、片親性ダイソミーが同定されない例については、最初にCDKN1Cの配列解析、次いでCDKN1Cの遺伝子標的型欠失/重複解析を検討すべきである。
- 染色体マイクロアレイ(CMA)
オリゴヌクレオチドアレイやSNPジェノタイピングアレイを用いた染色体マイクロアレイを用いることで、発端者の有する欠失や重複を検出することができる。
知的障害を有する発端者については、最初にこのCMAを検討すべきかもしれない。
検出しうる欠失のサイズを決める要素は、使用するマイクロアレイのタイプ、ならびに11p15.5領域のプローブの密度である[Kerenら2013,Baskinら2014,Russoら2016]。
SNPアレイ解析を用いれば、父性片親性部分ダイソミーの検出も可能である。
- 核型分析
11p15.5領域を含む逆位や転座の同定を目的とした核型分析も検討対象になりうる。
なお、こうしたタイプのものは、BWS罹患者の1%未満である。
- マルチ遺伝子パネル
CDKN1Cその他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルも検討対象になりうる。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
したがって、どのマルチ遺伝子パネルを用いれば、意義の不確かなバリアントや現状の表現型と無関係な病的バリアントの検出を抑えつつ、いま問題にしている疾患の遺伝的原因を最も安価に特定できる可能性が高いかという点について、臨床医の側であらかじめ検討しておく必要がある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:Beckwith-Wiedemann症候群で用いられる遺伝学的検査
| 手法 | 検出される病的バリアント/変化 | 変化の検出されるBWS罹患者の割合1 |
|---|---|---|
| メチル化解析2 | IC2の脱メチル化(母性) | 50%3 |
| IC1のメチル化(母性) | 5%3 | |
| IC2の脱メチル化プラスIC1のメチル化(父性片親性ダイソミー) | 20%3 | |
| 配列解析/遺伝子標的型欠失/重複解析4,5 | CDKN1Cの母性病的バリアントのヘテロ接合 | BWSの家族歴陰性例の5%6 |
| BWSの家族歴陽性例の40%近く6 | ||
| 核型分析 | 11p15.5領域の細胞遺伝学的な重複・逆位・転座 | 1%未満7 |
| マイクロアレイ(SNPベース) | 微小欠失,微小重複,父性片親性ダイソミー8 | 9%近く9 |
- BWSの臨床診断基準を満たした例について、遺伝子/座位別、表現型別、集団別、検査手法別に分類したときの罹患者の割合。
注:出現頻度は集団ごとに異なる[Sasakiら2007]。 - メチル化感受性を高めたアッセイ(例えば、MS-MLPA法、MS-qPCR法、サザンブロッティング法)を用いることで、11p15.5領域のエピゲノムやゲノムの変化を検出することが可能である。
メチル化感受性アッセイでは、以下のものを識別することができる。
―微小欠失や微小重複
―DNAメチル化の変化
―片親性ダイソミー(UPD)
メチル化データの解釈にあたっては、核型分析の結果を頭に入れておく必要がある。
というのは、父母の相対的な寄与割合に変化を及ぼすような核型異常(例えば、父性の重複)があると、メチル化異常という結果になって現れるからである。
11p15.5領域のUPDを確認するその他の手法としては、ショートタンデムリピート解析(STR解析)やSNP解析がある[Kerenら2013]。 - Bliekら[2001],Weksbergら[2001]
- 配列解析を行うことで、benign、likelybenign、意義不明、likelypathogenic、pathogenicといったバリアントが検出される。
病的バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、遺伝子の一部、1つの遺伝子全体、複数の遺伝子といった単位の欠失/重複については検出されない。
配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。 - 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。
具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなどがある。 - CDKN1Cの配列解析の検出率は、家族歴の有無により変わってくる[Hatadaら1997,Leeら1997,O‘Keefeら1997,Lamら1999,Liら1998,Brioudeら2015]。
- Slavotinekら[1997],Liら[1998]
- 父性片親性ダイソミーは接合後の体細胞性の組換えによって生じる。そのため、これは発端者サンプルのSNP解析でのみ検出可能である。
- Baskinら[2014]
臨床的特徴
臨床像
Beckwith-Wiedemann症候群(BWS)は、新生児低血糖、巨軀、巨舌、半側過形成、臍帯ヘルニア、胎児性腫瘍(例えば、Wilms腫瘍,肝芽腫,神経芽腫,横紋筋肉腫)、内臓巨大症、副腎皮質細胞腫大、腎異常(例えば、髄質異形成,腎石灰化症,髄質海綿腎,腎肥大)、外耳の皺/瘻孔などがさまざまな幅で現れる成長障害である。
BWSは1つの臨床的スペクトラムとみなすべきものであり、そこに属する罹患者の中には、特徴的臨床症候を数多く有する例もあれば、1つか2つしか有しない例もみられる。
Beckwith-Wiedemann症候群(BWS)にみられる特異的臨床症候それぞれの出現率は、報告により大きく異なっている。
その理由の一端は確認バイアスにある。
ただ、以下に述べるようなものは、明確に表現型の一部であると言うことができる。
出生前ならびに周産期
羊水過多、早産、胎児性巨軀の出現率は、50%に及ぶ模様である。
多くみられるその他の症候としては、過長臍帯、ならびに、平均で胎齢別の平均重量の2倍近くにも及ぶ胎盤肥大がある。
間葉性異形成胎盤として報告された児で、その後、BWSの症候を有していることが判明した例がみられる[Wilsonら2008]。
BWS乳児については、主として早産、巨舌、低血糖の合併症、そして稀には心筋症の合併症による死亡リスクの高まりがみられる。
それでも、以前に報告された20%という死亡率は、症候群の同定・治療に関する近年の進歩に鑑みると、過大な数字であるように思われる。
代謝異常
新生児低血糖については詳しく報告されており、BWS乳児の約50%にこれが生じる[Mussaら2016a]。
未発見ないし未治療のまま経過すると、発達上の後遺症をきたす重大なリスク要因となる。
大多数の例で、低血糖は軽度かつ一過性であるが、重症例では低血糖が続いていくことがある。
遅発性の低血糖(生後1ヵ月以内)も時折みられる。
それほど多くはみられないその他の内分泌/代謝/血液症候としては、甲状腺機能低下、高脂血症/高コレステロール血症、赤血球増多症などがある。
BWSの子どもでは、腎異常がなくても、高カルシウム尿症がみられることがある。
また、超音波検査で、BWS罹患者の22%に腎石灰化症が確認されている。
なお、一般集団での出現率は7%-10%である[Goldmanら2003]。
成長
巨舌(90%近くにみられる)や巨軀(50%近くにみられる)は、出生の段階で認められるのが普通であるが、両症候とも、出生後の発症例もみられる[Chitayatら1990,Brioudeら2013,Ibrahimら2014,Mussaら2016b]。
BWS罹患者の大多数は幼児期に急速な成長を示すものの、身長はふつう正常上限内に収まる。
成長速度は、通常、7-8歳前後には鈍化するようである。
半側過形成*は、通常、出生時には認められ、その後の成長とともに、より顕著になることもあれば、より目立たなくなることもある。
半側過形成は、身体の一部分に分節状に現れることもあれば、特定の器官・組織に現れるようなこともある。
複数の分節にこれが現れる場合、過形成が体の一側(同側)に限定されることもあれば、ある分節に生じた側とは逆の側(対側)に現れるようなこともある[Hoymeら1998]。
*注:半側過形成とは、非対称な過成長を生じるような細胞増殖の異常をいう。
BWSの場合、以前に用いられた片側肥大(こちらは細胞の大きさの拡大を指す用語である)に代わって、片側過形成(こちらは細胞数の増加を指す用語である)という表現が用いられるようになっている。
新生物
BWSの子どもは、新生物、特にWilms腫瘍や肝芽腫を原因とする死亡に対して高リスク状態にあるが、神経芽腫、副腎皮質癌、横紋筋肉腫についても同じ状態にある。
その他にも、悪性、良性を問わず、多種多様な腫瘍がみられる[Cohen2005]。
BWSの子どもの推定腫瘍発生リスクは7.5%で、範囲は4%から21%の間と推定されている[Cohen2005,Tan&Amor2006,Mussaら2016a]。
新生物に対するリスクの高まりは、0-8歳の間に集中しているように思われる。
ただ、多くはないものの、8歳を超えた年齢で腫瘍の発生をみた罹患者の報告例もみられる。
軽度の表現型(例えば巨舌と臍ヘルニア)を示す子どもの場合は、ことによると体細胞型のBWSである可能性もある。
しかし、その場合でも、BWSに関連する腫瘍発生に関して、(一般集団に比べ)高リスク状態にある可能性がある。
その理由の一部は、BWS関連の分子的変化がモザイク―BWS関連変化を有する細胞の多くが腫瘍発生に関して高リスク状態の器官(例えば、肝や腎)に存在する一方で、臨床症候として現れる組織には存在しないという状況―で存在するといったところにある。
BWSの表現型スペクトラムにあって最小の症候しか有しない子どもを評価する際には、常に疑いの目をもって、診断を確認するためにできるだけ遺伝子検査を用いようとする姿勢が求められる。
その他の器官系
- 前腹壁欠損
臍帯ヘルニア、臍ヘルニア、腹直筋離開などの前腹壁欠損が多くみられる。
- 口蓋裂
BWS罹患者のごく少数に口蓋裂がみられるが、これはふつう、CDKN1Cの母性継承の病的バリアントのヘテロ接合を有する場合に生じる[Hatadaら1997,Liら2001,Mussaら2016c]。
- 心血管系の問題
BWSでみられる心血管系の問題に関する報告の多くは、BWSの病原との関連を明確にした報告にはなっていない。
心拡大が罹患者の20%にみられる[Pettenatiら1986]とされ、これは乳児期に胸部X線写真を撮れば見つかる可能性があるものながら、ふつうは治療を行うことなく改善する。
- 心筋症の報告がみられはするものの、稀である。
- 11番染色体と17番染色体の間の均衡型転座によりKCNQ1に断裂をきたしたBWSの子どもの1例で、QT延長症候群がみられたとする報告が存在する[Kaltenbachら2013]。
- 腎奇形
腎奇形としては、髄質異形成、集尿系の重複、腎石灰化症、髄質海綿腎、嚢胞性変化、憩室形成、腎肥大などがある[Choykeら1998,Borerら1999,Mussaら2012]。
- 難聴
BWS罹患者における難聴の報告は稀で、感音性[Kantaputraら2013]、アブミ骨固着に起因する伝音性[Hopsuら2003]の両方の報告がある。
注:BWS罹患児の親のほうから、難聴や筋緊張低下に関する懸念が寄せられることも時にあるものの、こうした問題をはじめとする諸問題について、一般集団に比べBWS罹患者で高い出現頻度を示すかどうかという点の確認は困難である。
- 脳の異常
後頭蓋窩関連の脳の異常は、これまでわずかしか報告されていない[Gardinerら2012,Brioudeら2015]。
発達
染色体異常、脳奇形、または低酸素症や加療することなく放置された重症の低血糖といった既往がある場合を除き、ふつう、BWS児の発達は正常である。
親からの訴えが元となってBWSが確認されるに至った子どもについては、自閉症スペクトラム障害をはじめとする神経行動学的問題の発生頻度が高いとの報告がみられるものの、BWSにおけるそうした問題の頻度を論じるためには、正式な神経発達評価も含めたさらなる研究が必要である。
成人期について
小児期を過ぎると、一般に予後は良好である。
ただ、腎の髄質異形成や男性不妊などの合併症の可能性はある。
こうした問題は、特定の分子的サブタイプにおいて生じるものである可能性がある[Greerら2008]。
分子メカニズムごとにみた表現型との相関
分子メカニズムごとにみた一般的な表現型との相関を以下に示す。
ただ、どのBWS罹患者についても言えることであるが、分子的変化を元に、臨床症候の現れ方を正確に予測することはできない。
BWS罹患者における分子的変化以外の変動要因としては、体細胞モザイク、遺伝的背景、その他未知の要因などの関与が考えられよう。
新生物
- 11p15.5の片親性ダイソミーやIC1のメチル化がみられる例では、Wilms腫瘍や肝芽腫のリスクが最も高くなる。
- IC2の脱メチル化がみられる例では、腫瘍発生リスクが低く、今日までにWilms腫瘍の報告はみられない。
- 母由来のCDKN1Cアレルに現れた遺伝子内バリアントについては、以下のものとの関連がわかっている。
- 神経芽腫の症例の中の少数[Bliekら2001,Weksbergら2001,DeBaunら2002,Rumpら2005,Alsultanら2008,Kuroiwaら2009]
- 子どもにみられた神経節芽腫、急性リンパ性白血病、神経芽腫、成人でみられた黒色腫の各1例[Brioudeら2015]
注:白血病や黒色腫は、BWSを有しない例においても一定の頻度で現れることを考えると、こうした例が、互いに無関係な2つの疾患がたまたま同時に生じたものか、それともこうした悪性腫瘍が実際にBWSに起因して生じたものかという点については、判断の難しいところである。
半側過形成
半側過形成は、11p15.5の父性片親性ダイソミーのモザイクで最も高頻度にみられるが、IC2やIC1の分子的変化を有する例においてもみられる[DeBaunら2002,Shumanら2002,Enklaarら2006,Ibrahimら2014,Mussaら2016b]。
陽性の家族歴
CDKN1Cのヘテロ接合性病的バリアント、IC1の欠失、IC2の重複(これは稀)の場合に、家族歴が陽性となりやすい[Weksberg&Shuman2004,Cooperら2005,Prawittら2005,Enklaarら2006,Percesepeら2008,Scottら2008b,Bliekら2009]。
口蓋裂
口蓋裂は、母由来のCDKN1Cアレルのヘテロ接合性病的バリアントに関連して生じる[Hatadaら1997,Liら2001]。
臍帯ヘルニア
臍帯ヘルニアは、主として、IC2の変化や、母由来のCDKN1Cアレルのヘテロ接合性病的バリアントに関連して生じる[Ibrahimら2014,Brioudeら2015,Mussaら2016a,Mussaら2016c]。
巨舌と巨軀
巨舌や巨軀は、分子レベルのすべてのサブタイプでみられる顕著な症候である[Ibrahimら2014,Mussaら2016b]。
脳奇形
後頭蓋窩を中心とした脳奇形は、IC2の分子的変化や、母由来のCDKN1Cアレルのヘテロ接合性病的バリアントに関連して生じる[Gardinerら2012,Brioudeら2015]。
発達遅滞
発達遅滞は、細胞遺伝学的分析で検出可能な父由来の11p15.5の重複に関連して生じる[Slavotinekら1997]。
重度の表現型
BWSの重度の表現型は、11p15.5の片親性ダイソミーに関する高レベルの体細胞モザイクに関連して生じる[Smithら2006]。
女性の一卵性双生児
BWSの女性一卵性双生児不一致例は、IC2の脱メチル化に関連して生じるもののようである。
男性の一卵性双生児の誕生は女性よりずっと少ないが、こちらのほうはさまざまな分子的変化に関連して生じている[Weksbergら2002,Smithら2006]。
不妊
生殖補助医療(ART)を行った場合も行わなかった場合も含め、不妊症は、IC2の脱メチル化に起因するBWS児の発生頻度の高まりと関連があるように見受けられる[DeBaunら2003,Gicquelら2003,Maherら2003a,Maherら2003b,Hallidayら2004]。
浸透率
インプリンティング領域の親起源効果を考えたとしても、家族性の例における浸透率は、かなり高いレベルにある。
親起源効果というのは、例えば、CDKN1Cに病的バリアントが存在したとしても、それが父由来のアレルに生じたものの場合は、通常、発現に至らない(すなわち、正常なインプリンティングプロセスにより、その病的バリアントにはサイレンシングが生じている)ため、BWSの症候が現れないといったことである。
命名法について
BWSは当初、臍ヘルニア(exomphalos)、巨舌(macroglossia)、巨軀(gigantism)の3症候の頭文字をとって、EMG症候群と呼ばれていた。
発生頻度
10,000人に1人[Mussaら2013]から13,700人に1人[Thornburnら1970]といった報告にある発生頻度の数字は、表現型が軽微であるため診断に至っていない例が存在することを考えると、過少な推定値であるように思われる。
BWSは広い範囲の民族集団で報告されており、男女比は1:1である。遺伝子の上で関連のある疾患(同一アレル疾患)
見かけ上、孤発性と思われる半側過形成の罹患者に、IC2の脱メチル化、IC1のメチル化[Martinら2005]、11p15.5の父性片親性ダイソミー[Shumanら2002]といった、11p15.5領域の分子的変化がみられたとする報告がある。
そうした例の一部は、BWSの臨床的スペクトラムの中の軽度側の端にある例と思われるが、その他に、これ以外の分子的変化の体細胞モザイクによって生じたものであろうと思われる例も存在する。
孤発性のWilms腫瘍の中には、IC1の高メチル化、11p15.5の父性片親性ダイソミー、欠失や挿入などの11p15.5領域のゲノム異常など、11p15.5領域の構造的変化に起因して生じる例がみられる[Scottら2008a]。
父性のIC1の脱メチル化の体細胞モザイクによりSilver-Russell症候群や孤発性の半側低形成が生じることがある[Zeschnigkら2008,Eggermann2009,Eggermannら2015]。
サイクリン依存性キナーゼ阻害因子の安定性亢進をもたらす母性継承性のCDKN1Cの病的バリアントが、Silver-Russell症候群の1家系で報告されている[Brioudeら2013]。
また、CDKN1Cの機能獲得型ヘテロ接合性病的バリアントの母性継承により、IMAGe症候群が生じる[Arboledaら2012,Milaniら2014]鑑別診断
過成長
Beckwith-Wiedemann症候群(BWS)は、過成長を呈する子どもの鑑別診断に際して、しばしば検討対象となる。
BWSとの鑑別が必要な過成長症候群には、今もって分類がなされていないものが存在することに注意が必要である。
BWSと発達遅滞の両方を有するように見受けられるものの、染色体検査の結果が正常で、低酸素症や低血糖症の既往がない子どもについては、発達遅滞を生じる他の原因を検討する必要がある。
心臓の構造異常や伝導欠損がみられる場合は、Simpson-Galabi-Behmel症候群やCostello症候群も鑑別診断の対象になりうる。
表2:Beckwith-Wiedemann症候群(BWS)との鑑別診断において考慮すべき過成長疾患
| 疾患名 | 遺伝子 | 遺伝形式 | 臨床症候 | |
|---|---|---|---|---|
| BWSと重なる症候 | BWSと異なる症候 | |||
| Simpson-Galabi-Behmel症候群 1型 | GPC3 GPC4 |
XL |
|
|
| Perlman症候群 (OMIM267000) |
DIS3L2 | AR |
|
顔面の症候(小下顎症,耳介低位,低い鼻梁,逆V字形上赤唇)
|
| Costello症候群 | HRAS | AD2 | 新生児期はBWSに類似することあり(巨軀を有する場合)。 |
|
| Sotos症候群 | NSD1 | AD2 | 巨軀3 |
|
| ゲノムワイド父性片親性アイソダイソミーモザイク4 | 多数の遺伝子 | 散発性 |
|
|
AD=常染色体顕性;AR=常染色体潜性;XL=X連鎖性
- vanEeghenら[1999]
- 大多数の発端者は新生の病的バリアントに起因するもの。
- 巨軀は認めるものの、BWSに特徴的な他の症候がみられないといった場合は、NSD1のヘテロ接合性病的バリアントに関する検査を検討すべきである。
- Inbar-Feigenbergら[2013],Kalishら[2013]
半側過形成,分節状過成長
半側過形成や分節状過成長は、単発の症候として現れることがあり、また、Proteus症候群、PTEN過誤腫症候群、Klippel-Trenauney-Weber症候群(OMIM149000)、神経線維腫症1型など、他の症候群に伴って現れることもある[Hoymeら1998]。
顔や胸などの非対称については、斜頭症や胸壁変形の可能性を排除するための評価が必要となる。
臨床的マネジメント
最初の診断に続いて行う評価
Beckwith-Wiedemann症候群(BWS)と診断された罹患者については、疾患の範囲やニーズを把握するため、次のような評価を行うことが推奨される。
- 巨舌のみられる場合は気道が十分かどうかの評価。
- 巨舌により明らかな摂食障害がみられる場合は、摂食の専門職による評価。
- 新生児については、低血糖の評価。
低血糖が生後数日を超えて遷延する場合は、小児内分泌内科医による評価。
- 臓器肥大、器質的異常、腫瘍に関する評価を目的とした腹部超音波検査。
- これから外科的処置を行うというとき、あるいは、臨床評価で心臓の異常が疑われた場合は、心電図や心エコーを含む包括的な心評価。
- 肝芽腫の評価を目的として、最初の診断時にαフェトプロテインの定量。
特に、ごく幼い乳児については、結果の解釈の上で、該当する週齢に応じた正常値を利用することが重要である。
- 臨床遺伝医ないし遺伝カウンセラーとの面談
症状に対する治療
以下のような形にするのが適切である。
- 中枢神経系の合併症のリスクを低減するため、低血糖の治療を迅速に行う。
低血糖の始期は、時として数日程度、あるいは数ヵ月遅延することがあるため、両親には低血糖の症状に関する情報提供をきちんと行い、適切な医療の手配を両親のほうから求めることができるような態勢にしておく。
- 臍帯ヘルニアに対しては、生後ほどない時期に腹壁の修復術を行う。
ふつう、この手術は問題なく行えることが多い。
- 巨舌に起因して生じる障害に対し管理を行う。
- 気管内挿管が困難になることが予測される[Kimuraら2008]。
- 呼吸機能の評価を行う。
おそらくは、睡眠時無呼吸の可能性を考えて睡眠検査を行うといったことが中心になるものと思われる。
- 口蓋裂用の長めの特殊乳首を使用する等の摂食障害に対する管理を行う。
稀ながら、短期間だけ経鼻胃管栄養が使用されるようなこともある。
- BWSの自然経過に明るい形成外科医、矯正歯科医、言語治療士等で構成される頭蓋顔面チームによるフォローアップを行う。
舌の成長は経時的に鈍化する一方で、拡大した舌の体積に合うように、顎の成長は加速していく。
罹患児の中には舌の縮小術が有益と思われる例もみられはするものの、舌の縮小術で減少するのは舌の長さであって厚みのほうではない。
そのため、術後に美容的な問題や言語の問題が残って、その後も何らかの評価や治療が必要になるようなことがある[Tomlinsonら2007]。
- 小児期後期から思春期に、必要に応じ、矯正歯科的介入を行う。
- 言語障害に関する評価を行う。
- 標準的プロトコルに従って、口蓋裂の管理を行う。
- 顔面の片側過形成が顕著な場合は頭蓋顔面外科医への紹介を行う。
- 半側過形成により下肢長の顕著な左右差がみられる場合には、整形外科医との相談を行う。
- 長いほうの脚の成長板を閉鎖させることで最終的な下肢長の均等化を図る手術は、思春期の早い段階で行う必要があろう。
- 小児腫瘍学のプロトコルに従って、新生物の治療を行う。
- BWS罹患者の中には、カルシウムの排泄・沈着の亢進(すなわち、腎石灰化症)の背景に尿路奇形が存在する例がみられる。
- 腎超音波検査でカルシウム沈着が判明した罹患者については、高カルシウム尿症の評価や腎のCTスキャンが有用な場合がある。
- ◦尿中カルシウムの上昇や器質的な腎奇形が同定された場合は、小児腎臓内科医への紹介を行う。
- 消化管の器質的異常を有する子どもについては、関連する専門分野の医師に紹介する。
- 標準的プロトコルに従って、心臓の問題の管理を行う。
- 乳児発達刺激プログラム、作業療法、理学療法などによる標準的介入、併せて、発達遅滞を有する子どもに対しては、個別教育プログラムの策定を行う。
二次的合併症の予防
尿路感染症が疑われる場合には、腎への二次的なダメージを予防するため、評価を迅速に行って標準治療を行うことが適切である。
定期的追跡評価
以下のような形にするのが望ましい。
- 特に新生児期については、低血糖に関するモニタリングを行う。
- 胎児性腫瘍に関するスクリーニングを行う。これは従来、次のような内容を含むものとされている(「注:」も併せて参照のこと)。
- 8歳までは腹部超音波検査を3ヵ月ごとに行う[Beckwith1998,Tan&Amor2006,Clericuzio&Martin2009,Zarateら2009]。
- 肝芽腫の早期発見を目的とした血清αフェトプロテイン(AFP)濃度の測定を、満4歳になるまでは2-3ヵ月ごとに行う[Clericuzio&Martin2009]。
BWS児においては、生後1年間は血清AFP濃度が上昇することがある[Evermanら2000]。
AFPに上昇がみられ、画像診断で特段の疑わしい病変が見つからない場合は、1ヵ月後にAFP濃度を追跡測定するとともに、ベースラインの記録としての肝機能検査を行うことで、経時的な血清AFP濃度の傾向をみる基準とすることができる。
もしも濃度が低下していかないようであれば、隠れて存在する腫瘍を徹底的に探索するようにすべきである[Clericuzioら2003]。
注:(1)検出された分子レベルの変化の内容によって、腫瘍のサーベイランスに関するガイドラインに変化をもたせることを提唱する向きもある。
Scottら[2006]は、IC2の変化を有するBWS児については、Wilms腫瘍に関するスクリーニング不要ではないかと述べている。
Brioudeら[2013]は、IC2の脱メチル化を呈するBWS児に対する超音波の評価について、臨床診断の際に行った後は、内臓巨大症ないし「重度の」半側過形成がある場合にのみ超音波によるサーベイランスを行うこととし、それ以外の場合は臨床診査のみとすることを推奨している。
Mussaら[2016b]は、IC2の脱メチル化を有する罹患者に対して超音波によるサーベイランスや「腫瘍マーカー」のチェックを行うことに、どれほどの合理性があるか疑わしいとしている。
そして、その後、彼らが作成に携わったBWSに関するイタリア科学委員会のガイドラインの中で、彼らは、近い将来、臨床の場における腫瘍スクリーニングは行われなくなるであろうと述べている。
ただ、本GeneReviewの著者らは、個人的経験に基づいて、分子レベルの病因の如何にかかわらず、すべてのBWS罹患児に対し、腫瘍サーベイランスの推奨を続けている。
(2)神経芽腫のスクリーニングを目的として、定期的に胸部X線写真の撮影や尿中バニリルマンデル酸(VMA)、ホモバニリン酸(VHA)の定量を行うことがこれまで提唱されてきたが、それにより得られる利得が少ないため、多くのスクリーニングプロトコルには、項目として組み込まれていない現状がある。
- 8歳から思春期中盤にかけての時期に、腎石灰化症や髄質海綿腎の精査を要する例を特定する目的で、腎超音波検査を年に1度実施する。
結果が陽性であった場合は、腎臓内科医に紹介して、その後の評価や経過観察を行ってもらう。
成人期における腎疾患の自然的経過に関する評価は、今のところ存在しない。
成人期以前には特段の所見なく、成人期になって発症する腎疾患というものも、可能性としては残っている。
したがって、成人期においても定期的に腎評価を続けることを積極的に検討すべきである。
- 超音波検査で特段の異常所見を示さないBWS罹患者で、時として尿中カルシウム/クレアチニン比が異常値を示す例がみられる。
そのため、BWS診断時以降、年に1-2度の割合で、尿中カルシウム/クレアチニン比の測定を行うことを検討する[Goldmanら2003]。
- 子どもに対するルーチンのケアの一環として、発達スクリーニングを行う。
リスクを有する血縁者の評価
BWS罹患者の同胞については、予防処置をできるだけ早期に開始することで利益が得られる人を特定する意味合いから、新生児の段階で評価を行うことが望ましい。
評価の内容は以下の通りである。
- 母性のCDKN1Cの病的バリアント、ないし、11p15.5領域の家族性の重複、欠失、細胞遺伝学的に視認可能な変化等がすでに判明している場合は、遺伝学的検査を行う。
- 仮に、出生前の検査で低血糖を示す明らかな臨床所見がみられなかった場合でも、リスクを有する新生児の同胞に対しては、低血糖に関するモニタリングを行う。
- BWSに関し一卵性双生児不一致例であった場合、胎児循環を共有していたこと、ならびに体細胞モザイクの可能性を考慮し、一見したところ非罹患児と思われるほうの児についても、腫瘍サーベイランスを行うことを前向きに検討する。
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Beckwith-Wiedemann症候群(BWS)は、BWSクリティカル領域にあるインプリンティングが施された遺伝子に、いくつかある遺伝的メカニズムの1つが作用して発現異常をきたすことで生じるものである。
家族構成員のリスク
発端者の両親
- 大多数のBWS罹患者について言うと、その親は罹患者ではない。
- 家族歴のわかっていないBWS罹患児の両親に対する評価は、幼少期にBWS関連の病歴・家族歴が存在したかどうかといった点の評価が中心となる。
乳児期から小児期にかけての写真が有用なこともある。
成人となった現状にあっては、身体的診査の意義は限定的であるものの、耳瘻孔/耳垂の皺が残存している場合がある。
ただ、そうした皺は、一般集団でもそれほど珍しいものではない。
- メチル化異常は、背後にある微小欠失や微小重複が原因となって生じるという性質のものではないため、通常、継承されるものではない。
したがって、両親に対してメチル化異常の検査を行っても、通常は「異常なし」という結果になる。
発端者の同胞
- BWS児の同胞の有するリスクは、発端者の有するBWSの遺伝的背景によって変わってくる(表3)。
大多数の家系においては、再発リスクは1%未満であるが、病因によっては、50%の再発リスクとなる。
- 再発リスクの評価のために。
遺伝カウンセリングを行う上では、背景にある遺伝的メカニズムを同定しておく必要がある。
表3:家族歴と分子メカニズムを基礎としてみたときのBWS発端者の同胞の有するリスク
| 家族歴 | BWSの分子メカニズム | 発端者の同胞の有するリスク |
|---|---|---|
| 陰性 (正常核型) (85%近く) |
母性染色体のIC2の脱メチル化(50%) | ゲノム異常(例えば、微小欠失/重複[MS-MLPA法で確認]やNLRP2など別の座位の病的バリアント)がみられない場合は非常に低い。 |
| 不明(最大20%) | 不明ながら、経験的には低い1。 | |
| 11p15.5の父性片親性ダイソミー(最大20%) | 非常に低い。 それは、この領域のUPDが、接合後の体細胞性組換えに起因するものと思われるためである。 |
|
| 母性染色体のIC1のメチル化(5%) | ゲノムに異常がみられない(MS-MLPA法で確認)場合は非常に低い。 | |
| CDKN1Cの病的バリアント(最大5%) | 臨床的に非罹患者である母親から罹患者である子へ、CDKN1Cの病原性バリアントが継承された例が数例報告されている2,3。 こうした家系における再発リスクは、おそらく50%と思われる。 |
|
| 微小欠失/重複 | 50%以下。 ゲノム異常のためにメチル化の変化が生じているような場合は、たとえ家族歴が陽性でないように思われたとしても、両親に対する分子遺伝学的検査を検討すべきである4,5。 もし片親に同じゲノム異常がみられるようであれば、片親起源効果を考慮しなければならず、再発リスクは50%に達する可能性がある。 |
|
| 陽性 (正常核型) (15%近く) |
CDKN1Cの病的バリアントを発端者で検出 (40%) |
母親がCDKN1Cの病的バリアントを有していた場合、発端者の同胞の有するリスクは50%ということになる。 父親がCDKN1Cの病的バリアントを有していた場合、同胞に対するリスクは高くなるものの、その正確な数字については、経験値を明らかにするための今後の研究を待つ必要がある。 発端者でみられたCDKN1Cの病的バリアントを両親ともに有していなかった場合は、同胞に対するリスクは低い。 ただ、生殖細胞系列モザイクの可能性は残っている。 |
| CDKN1Cの病的バリアントが発端者で非検出 (最大60%) |
50%以下。 | |
| 微小欠失/重複 | 50%以下。 ゲノム異常のためにメチル化の変化が生じているような場合は、たとえ家族歴が陽性でないように思われたとしても、両親に対する分子遺伝学的検査を検討すべきである4,5。 もし片親に同じゲノム異常がみられるようであれば、片親起源効果を考慮しなければならず、再発リスクは50%に達する可能性がある。 |
- ここに入る罹患者の一定割合、特に半側過形成を呈する例については、11p15の片親性ダイソミーモザイクである可能性がある。
- Hatadaら[1997],O‘Keefeら[1997],Lewら[2004]
- あまりないことと思われるが、臨床的に非罹患者と思われた父親から、CDKN1Cの病的バリアントの父性の伝達を受けた1例が報告されている[Leeら1997]。
- メチル化異常の中には、高解像度核型分析では視認不能な微小欠失ないし微小重複に伴って生じるものがある。
- 家族性の例で、CDKN1Cの病的バリアントが検出されず、核型も異常なしといった場合には、微小欠失/微小重複の継承を調べる検査の実施を検討すべきである[Niemitzら2004,Sparagoら2004,Prawittら2005]。
その場合、採取した組織中のモザイクのレベルが低いと、これが検出されない可能性がある。
したがって、別の組織(例えば、過形成側の皮膚)を用いた検査の追加を検討すべきである。
そのため、メチル化異常がみられる場合には、少数とはいえ、再発リスクが顕著に高まるこうした家系を特定するために、染色体マイクロアレイを用いる必要がある。
発端者の子
表4:家族歴と分子メカニズムを基にしてみたときのBWS発端者の子の有するリスク
| BWSの分子メカニズム | 発端者の子の有するリスク |
|---|---|
| IC2の脱メチル化 | 生殖細胞系列では、インプリンティングの状態はリセットされる。そのため、ゲノム異常がない場合のリスクは低い。 ただ、経験的なリスクを数字で示したデータはまだ存在しない1。 |
| IC1のメチル化 | 生殖細胞系列では、インプリンティングの状態はリセットされる。そのため、ゲノム異常がない場合のリスクは、理論上は低い。 ただ、経験的なリスクを数字で示したデータはまだ存在しない。 |
| CDKN1Cの病的バリアント | 女性発端者の場合は50%。 男性発端者の場合は50%を下回るが、リスクを数字で示すには、報告例が少な過ぎる状況である。 |
| 11p15.5の父性片親性ダイソミー | 非常に低いと思われる。 ただ、経験的なリスクを数字で示したデータはまだ存在しない。 |
| 11p15.5の微小欠失/微小重複 | 50%2。 子に生じる表現型は、伝達元である親の性別によって、BWSかSilver-Russell症候群、どちらかの形で現れる3。 |
- Niemitzら[2004]
- 家族性の例として、IC1の微小欠失や、稀ではあるがIC2の微小重複の報告が存在する[Hatadaら1997,Leeら1997,Sparagoら2004,Weksberg&Shuman2004,Cooperら2005,Prawittら2005,Enklaarら2006,Percesepeら2008,Scottら2008b,Bliekら2009]。
- Sparagoら[2004]
他の家族構成員
他の血族の有するリスクは、分子レベルでの病因によって変わってくる。
発端者の染色体に異常がみられる場合の血縁者の有するリスク
発端者の親
発端者の染色体構成に均衡型ないし不均衡型転座がみられる場合、その親は、均衡型の染色体再配列を有しているリスクがある。
そのため、親に対して染色体分析を行うべきである。
染色体異常を有する発端者の同胞
- 同胞の有するリスクは、両親の細胞遺伝学的所見によって変わってくる。
- 母親が11p15の均衡型転座を有していた場合、同胞への再発リスクは50%ということになり、BWSの表現型は、母親の有するこの転座の伝達に従った分離を示すものと思われる。
- 父親が11p15領域を含む均衡型転座を有しており、子に11pの重複という形ですでに現れている場合、同胞への再発リスクは高まることになるものの、その正確な数字についてはわからない。
染色体異常を有する発端者の子
発端者が女性である場合、子の有するリスクは50%となる可能性がある。
男性発端者の場合、リスクは低いものと思われる。
表5:細胞遺伝学的異常を有するBWS発端者の同胞や子の有するリスク
| 細胞遺伝学的異常 | 発端者の同胞の有するリスク | 発端者の子の有するリスク |
|---|---|---|
| 細胞遺伝学的に検出された母性の11p15の転座ないし逆位 | 伝達元となるのが母親のほうであれば、50%となる可能性あり。 | 伝達元となるのが母親のほうであれば、50%となる可能性あり。 |
| 細胞遺伝学的に検出された父性の11p15の重複 | 評価不能 | 評価不能 |
発端者の他の血族
染色体異常が検出された場合は、常に、リスクを有する他の血族に対し、その状況を明確化するための染色体検査を行うべきである。
関連する遺伝カウンセリング上の諸事項
リスクを有する血族に対して、早期診断、早期治療を目的として行う検査関連の情報については、「管理」の中の「リスクを有する血縁者の評価」の項を参照されたい。
家族計画
- 遺伝的リスクの確定、出生前検査に関する遺伝カウンセリングといったことに最も適しているのは、妊娠前の時期である。
症候が現れていないもののリスクは有している血族の遺伝的状況を明らかにするための検査を受けるかどうかを決断するのに最も適した時期は、妊娠前の時期である。
- 罹患者である若い成人、あるいはリスクを高めるようなゲノムの変化を保有している若い成人(例えば、父親から11p15の均衡型転座を継承している正常女性)に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
生殖補助医療(ART)に伴って生じる可能性のあるインプリンティング障害のリスク
不妊症/ARTとインプリンティング障害との関連性を疑わせるデータが出ている[DeBaunら2002,Maherら2003a,Maherら2003b]。
より最近のものでは、DeBaunら[2003],Gicquelら[2003],Maherら[2003a],Hallidayら[2004]が、インプリンティングがなされた11p15のセントロメア側にあるIC2におけるインプリンティングの変化を助長し、その結果として、不妊症のカップルの子どもや、ARTを受けた女性の子どもにBWSの発生率が高まる可能性があるというデータを報告している。
こうしたインプリンティング異常が生じる上で、不妊症、ならびにそれと対を成すARTの背景にあるものが、いかなる役割を果たしているのかという点については、よくわかっていない。
胚発生における着床前期という時期は、インプリンティングを維持する上で重要な時期である。
今のところ、ARTの手順の中の何か特定のものがBeckwith-Wiedemann症候群のリスクを高めているというデータはないものの、ARTの際の数々の手順、例えば、卵胞刺激のプロトコル、生物学的手技、配偶子の成熟ステージ、培養に用いる培地、胚を移すタイミングといったものがインプリンティングに影響を及ぼしている可能性がある。
ART関連のAngelman症候群の報告もみられることから、発生初期に生じるエピゲノム関連のエラーは、何もBeckwith-Wiedemann症候群に限ったことではないと言えそうである。
これらはいずれも後ろ向き研究のデータであるが、こうしたデータがはっきりと指し示していることは、今後、ARTで誕生した子どもを追跡していくことが必要であるという点、ならびに、インプリンティングエラーの発生リスクの上昇がARTに起因するものであるか否か、もしそうであるとすると、その原因はARTだけにあるのか、それとも背景にある親の側の不妊症との絡みでそうなるのかといったことを明らかにしていくため、より大規模な前向き研究が必要であるという点である[Debaunら2003,Gicquelら2003,Gosdenら2003,Maherら2003a,Weksbergら2003,Schieveら2004,Weksbergら2005]。
一卵性双生児の誕生
BWSに関して不一致の一卵性双生児(通常は女性)は、同時に、皮膚線維芽細胞におけるIC2の脱メチル化についても不一致であるが、血液細胞についてみると、幅をもちつつも一致している。
これはおそらく、胎児循環を共有していたためと思われる[Weksbergら2002]。
男性の一卵性双生児の出現頻度は女性よりずっと低く、分子レベルでは11p15領域の片親性ダイソミーやIC1のメチル化といった所見を示す。
こうした双生児については、同胞に再発がみられたとする報告が存在しないため、再発リスクについてはよくわかっていない。
DNAバンキング
DNAバンキングとは、将来の利用に備えてDNA(通常、白血球から抽出したもの)を保存しておくことをいう。
検査の手法であるとか、遺伝子・アレルのバリアント・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。
そのため、分子診断の確定していない(すなわち、背景にある病原のメカニズムが未解明の)発端者については、DNAの保存を検討すべきである。
出生前検査
家族歴陽性の場合
すでにBWSの子どもがいる家族については、もし出生前診断に関心があるのであれば、いくつもの選択肢がある。
妊娠管理を念頭に出生前診断を希望する家族もいれば、堕胎を検討する家族もあろう。
発端者の有する細胞遺伝学的異常ないし分子遺伝学的異常の内容が判明している場合は、リスクを有する妊娠に対応した適切な検査を行うことが可能である。
11p15を含む重複/欠失、CDKN1Cの病的バリアントといったゲノムの変化については、絨毛採取(CVS)ないし羊水穿刺で得られたサンプルを用いて胎児DNAの解析を行うことで同定することが可能である。
羊水から抽出したDNAについては、これまでに偽陰性の報告がみられはするものの、それでも、現時点では、胎児のメチル化状態を評価する上で最も信頼度の高い組織サンプルであるように思われる[Eggermannら2015]。
羊水細胞培養の示すところはクローン化した所見であって、胎児の状態を正確に反映したものではないことがあり、ちょうど出生後の検査で体細胞モザイクのサンプルを検査しているようなものである可能性がある。
これと同様に、CVSで採取した組織を用いてメチル化状態の出生前診断を行っても、信頼性のある結果は出ない可能性があり、実際に偽陽性の結果がみられたとの報告が存在する[Eggermannら2015]。
こうした問題がなぜ生じるかというと、胎盤発生初期においてメチル化状態が確立するタイミングの問題があるからである。
遺伝カウンセリングにあたっては、エピゲノムの変化に関する出生前検査の限界をきちんと押さえたものにする必要がある[Eggermannら2015]。
遺伝的メカニズムが判明しているか否かにかかわらず、BWSに関して高リスクの妊娠については、次のように言うことができる。
- 臍帯ヘルニアがある場合には、妊娠16週の段階で母体の血清αフェトプロテイン(AFP)濃度が上昇する可能性がある。
- 第2三半期後期において妊娠週齢比でみたときに亢進している成長パラメーターを評価するとともに、腹壁欠損、臓器肥大、腎奇形、口蓋裂、心奇形、巨舌を検出することを目的として、妊娠19-20週で超音波検査を行い、妊娠25-32週で再度これを行うという方法がある。
妊娠10-14週の間に行った超音波検査で胎児の後頸部の厚みの増大と臍ヘルニアがみられ、その後、BWSが判明した1例の報告がみられる[Soukaら1998]。
注:(1)超音波検査により胎児に奇形や成長異常がみられなかったとしても、BWSの臨床症候に一定の幅が存在する以上、BWSのリスクは依然として残ることになる。
(2)出生前検査で明らかな臨床所見がみられなかった場合でも、低血糖に関する新生児のモニタリングは行うべきである。
家族歴陰性の場合
BWSの家族歴はないものの、妊娠中の超音波検査で単発性の明らかな臍帯ヘルニアのような異常が同定された場合は、次のような追加の検査を検討すべきである[Porterら2009,Wilkins-Haugら2009]。
- 羊膜細胞を用いて行うメチル化状態の変化を調べるための分子遺伝学的検査。
メチル化状態に変化がみられなかった場合は、CDKN1Cの病的バリアントを調べる検査。
- 11p15のコピー数変化を調べるための染色体マイクロアレイ、ないし、11p15の重複、逆位、転座を評価するための細胞遺伝学的検査。
- 胎児の成長を評価するとともに、BWSに特徴的な他の異常を同定することを目的として一連の形で行う超音波検査。
注:妊娠週齢は、最後の正常月経開始日から数えた週数、あるいは、超音波の計測値を基に数えた週数で表現される。
BWSが強く疑われるときは、分子遺伝学的検査を行うことが可能である。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
-
Beckwith-WiedemannChildren'sFoundationInternational
www.beckwithwiedemann.org
・MedlinePlus
Beckwith-Wiedemannsyndrome
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Beckwith-Wiedemann症候群:遺伝子とデータベース
| クリティカル領域 | 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMG | ClinVar |
|---|---|---|---|---|---|---|
| BWS | 不明 | 11p15.4 | 不明 | |||
| CDKN1C | 11p15.4 | サイクリン依存性キナーゼ阻害因子1C | CDKN1Cdatabase | CDKN1C | CDKN1C | |
| H19 | 11p15.5 | 不明 | H19@LOVD | H19 | H19 | |
| IGF2 | 11p15.5-p15.4 | インスリン様成長因子Ⅱ | LOVD-Growthconsortium (IGF2) |
IGF2 | IFG2 | |
| KCNQ1 | 11p15.5 | 電位依存性カリウムチャンネルサブファミリーKQTメンバー1 | KCNQ1@LOVD KCNQ1@ZAC- GGM |
KCNQ1 | KCNQ1 | |
| KCNQ1OT1 | 11p15.5 | 不明 | KCNQ1OT1@ LOVD |
KCNQ1OT1 | KCNQ1OT1 |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Beckwith-Wiedemann症候群関連のOMIMエントリー(閲覧はすべてOMIMへ)
| 103280 | H19, IMPRINTED MATERNALLY EXPRESSED NONCODING TRANSCRIPT; H19 |
| 130650 | BECKWITH-WIEDEMANN SYNDROME; BWS |
| 147470 | INSULIN-LIKE GROWTH FACTOR II; IGF2 |
| 600856 | CYCLIN-DEPENDENT KINASE INHIBITOR 1C; CDKN1C |
| 604115 | KCNQ1-OPPOSITE STRAND/ANTISENSE TRANSCRIPT 1; KCNQ1OT1 |
| 607542 | POTASSIUM CHANNEL, VOLTAGE-GATED, KQT-LIKE SUBFAMILY, MEMBER 1; KCNQ1 |
| 616186 | H19/IGF2-IMPRINTING CONTROL REGION |
分子レベルの病原
インプリンティング
インプリンティングとは、ある1つの遺伝子を構成する2つのアレルのDNAが別個の修飾を受け、個々の遺伝子中の一方のアレルだけが片親特異的に発現するエピゲノムの仕組みをいう[Barlowら1994]。
インプリンティングを受けた遺伝子はゲノム中の特定のドメインに集中して存在する。
そして、生殖細胞系列を通じた伝達の過程で、密接につながった一群のインプリント遺伝子をリセットする作業の制御をインプリンティングセンターが担っている[Nicholls1994]。
遺伝子/座位
成長因子や腫瘍抑制遺伝子を含む11p15領域にマッピングされる数多くのインプリント遺伝子が、原因遺伝子として関与していることが明らかになっている。
そして、インプリンティングセンター(IC1とIC2)が、染色体の広いドメインにわたって遺伝子発現を制御している。
Beckwith-Wiedemann症候群(BWS)においては、この領域に数多くの分子的変化が生じている。
BWSに関連してみられるインプリント遺伝子の発現の変化
BWSクリティカル領域には、2つのインプリントドメインが存在する(図1a参照)。
ドメイン1はテロメア側にあり、ここにはH19、IGF2というインプリント遺伝子が存在する。
H19は非コードRNA、非翻訳RNAで、腫瘍抑制因子として機能している可能性がある。
一方、IGF2のほうは、強力な胎児性成長因子である。
H19とIGF2は相補的に発現するインプリント遺伝子であり、H19のほうは母性発現、IGF2のほうは父性発現を示す。
このドメインの発現制御は、H19の上流にあるインプリンティングセンター1(IC1)(メチル化可変領域1[DMR1]とも呼ばれる)により行われている。
IC1は通常、父性アレルではメチル化状態、母性アレルでは非メチル化状態にある。
転写の制御は、IC1内のコンセンサス配列にジンクフィンガーインシュレータータンパク質CTCFが結合することで行われる。
CTCFは、非メチル化配列(母性アレル)にのみ結合し、下流にあるエンハンサーがIGF2のプロモーターに対して作用するのを阻害する[Harkら2000]。
ドメイン2はセントロメア側に位置し、ここにはインプリント遺伝子CDKN1C、KCNQ1、KCNQ1OT1が存在する。
このドメインの制御は、インプリンティングセンター2(IC2)(以前はメチル化可変領域2[DMR2]と呼ばれていた)により行われている。
IC2は、KCNQ1のイントロン10内に存在する。
IC2[Smilinichら1999]内には、KCNQ1OT1のプロモーターが存在する。
なお、KCNQ1OT1は、制御機能を果たしていると考えられる非コードRNAである[Pandeyら2008]。
この領域の制御機構の詳細は不明であるが、母性染色体上のIC2脱メチル化により、通常であれば父性発現を示すKCNQ1OT1が両アレル性に発現する結果となることが知られている。
さらに、BWS罹患者のうち、母性染色体のIC2の脱メチル化を示す例については、CDKN1Cの発現低下に至ることがわかっている[Diaz-Meyerら2003]。
インプリンティングセンター(IC)
インプリンティングセンターとは、隣接する広い領域のインプリント遺伝子群の発現を、シス因子の形で制御するDNA領域をいう。
ICは通常、DNAメチル化の変化、ならびにヒストン修飾の変化といった形で機能し、インプリンティング制御領域(ICR)とか、メチル化可変領域(DMR)といった呼び方がなされることもある。
- IC1は、11p15.5のテロメア側にあるインプリンティングセンターで、H19プロモーターの上流にマッピングされており、H19とIGF2の両方を制御している。
ICR1、DMR1、H19DMRといった呼び方がなされることもある。
正常では、父性アレルのほうはメチル化、母性アレルのほうは非メチル化状態にある(すなわち、特異的メチル化状態)。
- IC2は、セントロメア側にあるインプリンティングセンターで、KCNQ1OT1、KCNQ1、CDKN1Cなど、いくつかの遺伝子を制御している。
ICR2、DMR2、KvDMR1といった呼び方がなされることもある。
正常では、母性アレルのほうはメチル化、父性アレルのほうは非メチル化状態にある(すなわち、特異的メチル化状態)。
- メチル化ないし高メチル化というのは、対照サンプルと比較して、DNAのメチル化の程度が高まることをいう。
インプリント領域について言うと、正常では非メチル化状態にあるアレルのメチル化という意味で用いられることがある。
- 脱メチル化ないし低メチル化というのは、対照サンプルと比較して、DNAのメチル化の程度が低下することをいう。 インプリント領域について言うと、正常ではメチル化状態にあるアレルの脱メチル化という意味で用いられることがある。
- 片親性ダイソミー(UPD)
BWS罹患者の20%に、11p15UPDの体細胞モザイクがみられる。
このUPDは、全例が、体細胞性の組換えイベントに起因して父性のアイソダイソミーが生じたという成り立ちのようである。
UPDの例の大多数は11p15の父性UPDの部分モザイクを示すことから、その背景にあるメカニズムは、接合後の体細胞性組換えイベントによって11p15のUPDモザイクが生じたというものであることが示唆される。
したがって、採取した組織中のモザイクのレベルが低ければ、UPDは検出されない可能性がある。
その場合、他の組織(例えば、皮膚線維芽細胞、腫瘍の生検)の検査を考慮する必要がある。
- IGF2
IGF2はインプリント遺伝子で、父性発現の胎児性成長因子をコードしている。
IGF2のインプリンティング異常により両アレル性の発現となっている状態が、一部のBWS罹患者、ならびに、Wilms腫瘍を含む複数の腫瘍で明らかになっている。
父性のigf2アレルに病的バリアントを有するマウスは小さく生まれる一方で、同じ病的バリアントが母性継承アレルに生じても、胎仔の成長に影響は及ばない。
また、igf2の過剰発現、ないし、Iigf2受容体の異常で、マウスの過成長が生じる。 - H19
これは母性発現遺伝子で、生物活性を有する非コードmRNAをコードしている。
この非コードmRNAは、腫瘍抑制因子として機能している可能性がある。
BWS罹患者の約50%は、組織中でIGF2の両アレル性発現を示しつつ、IGF2とH19が非連動の形で発現する。
すなわち、大多数でH19の母性片アレル性発現が維持される。
少数ながら、H19の発現やメチル化に変化がみられるBWS罹患者も報告されている[Joyceら1997,Sparagoら2004]。 - CDKN1C
これはp57KIP2タンパク質をコードしている。
このタンパク質は、サイクリン依存性キナーゼ阻害因子ファミリーの1つで、細胞増殖に対する負の調節機能を果たしている。
この遺伝子は、腫瘍抑制遺伝子と推定されると同時に、胎児の成長に対し潜在的な負の調節因子ともなっている。
こうした2つの機能を有していること、ならびに、この遺伝子の母性優先発現(父性アレルの転写の部分的抑制)がみられたことから、BWSの候補遺伝子であることが示唆されるに至った。
この遺伝子の病的バリアントは、罹患者の約5%で報告されている。
CDKN1Cの病的バリアントは、臍帯ヘルニア、口蓋裂、家族歴陽性の罹患者でより多くみられる。
ただ、目下のところは、BWSの垂直的伝達の全例がCDKN1Cの病的バリアントに起因するものであると言い切ることまではできない現状である[Hatadaら1997,Leeら1997]。 - KCNQ1
KCNQ1によってコードされるタンパク質は、カリウムチャンネルの一部を構成し、心臓不整脈症候群の中の少なくとも2つ、すなわちRomano-Ward症候群とJervellandLange-Nielsen症候群への関与が明らかとなっている。
この遺伝子は、大多数の組織(心臓を除く)において母性発現性で、選択的スプライシングにより4種の転写産物が産生される。
そのうちの2つは非翻訳性転写産物である。 - KCNQ1OT1
KCNQ1OT1は、KCNQ1のイントロン10に起源をもつアンチセンス転写産物である。
KCNQ1OT1の5'側にある特異的メチル化プロモーター領域(IC2)のインプリンティング消失が、BWS罹患者の50%にみられる[Bliekら2001,Weksbergら2001]。
その他のインプリント遺伝子
PHLDA2(IPL、HLDA2、BWR1Cの名でも知られる)とSLC22A18(TSSC5、BWR1A、ITMの名でも知られる)は、ともに11p15領域にあるインプリント遺伝子である[Qianら1997,Daoら1998]。
両者とも胎生期において母性優先の形で発現する遺伝子で、CDKN1Cのセントロメア側に位置する。
両遺伝子とも、BWSへの直接的関与が明らかになっているわけではないが、負の成長調節機能を有しているものと考えられている。
最近になって、PHLDA2が胎盤/胎児の正常な発生に重要な役割を果たしていることが明らかになっている[Dóriaら2010,Tunsterら2010]。
この領域の遺伝子発現量は、マウス胎仔の成長制御を行う上で重要である。
マウスモデルでは、Igf2の上向き調節とCdkn1c(p57Kip2)の下向き調節により、BWS類似の表現型が生じる[Casparyら1999]。
更新履歴:
-
Gene Reviews著者: Cheryl Shuman, MS, CGC, J Bruce Beckwith, MD, and Rosanna Weksberg, MD, PhD, FRCPC, FCCMG, FACMG
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2016.8.11. 日本語訳最終更新日: 2022.8.1.[in present]
原文 Beckwith-Wiedemann Syndrome
印刷用![]()