鎖骨頭蓋異形成スペクトラム障害
(CleidocranialDysplasiaSpectrumDisorder)
[Synonyms:CleidocranialDysostosis]
Gene Reviews著者: KarenMachol,MD,PhD,RobertoMendoza-Londono,MD,MS,andBrendanLeeMD,PhD
日本語訳者: 佐藤康守(たい矯正歯科)、森貞直哉(兵庫県立こども病院)
GeneReviews最終更新日: 2023.4.13. 日本語訳最終更新日: 2023.8.6.
原文: Cleidocranial Dysplasia Spectrum Disorder
要約
疾患の特徴
鎖骨頭蓋異形成(CCD)スペクトラム障害は、古典的CCD(頭蓋縫合閉鎖遅延・鎖骨の低形成ないし無形成・歯の異常の3徴候)から、軽度型CCD、さらには骨格症候を伴わない歯の異常のみのものに至るまで、臨床的連続性を示す骨系統疾患である。古典的CCDスペクトラム障害を有する例では、出生時、大泉門は異常に大きく開いた状態を呈し、これは生涯にわたって開存することがある。鎖骨の低形成のため、肩は細く、なで肩になり、両肩を正中で合わせられる場合がある。中等度の低身長がみられることが多く、大多数の罹患者は非罹患者の同胞より低身長となる。歯の異常としては、永久歯の萌出遅延、乳歯の脱落障害、過剰歯などがある。CCDスペクトラム障害の罹患者は、反復性の洞の感染症、反復性の耳の感染症とそれに続発する伝音性難聴、上気道閉塞といったことの発生に関し、高リスクとなる。知能はふつう正常である。
診断・検査
CCDスペクトラム障害の診断は、特徴的臨床所見・X線写真所見がみられること、ないし、分子遺伝学的検査でRUNX2のヘテロ接合性病的バリアントが同定されることをもって確定する。
臨床的マネジメント
症状に対する治療:
頭蓋冠の欠損が大きい場合には、鈍的外傷から頭部を保護する必要がある。高リスクの活動に際しては、ヘルメットの着用が考えられる。前額部の陥凹に対する美容外科的対応や、低形成の鎖骨に対する延長術が検討対象となる。頭蓋顔面ならびに歯に異常がみられるため、麻酔管理計画は注意深く進める必要がある。気道確保について耳鼻咽喉科医と綿密に打ち合わせるようにする。脊椎に異常を有する可能性を考慮し、脊髄幹ブロックなどの代替麻酔手段を検討する。骨密度が正常値を下回るときは、カルシウム・ビタミンD補充の治療を進める。乳歯の残存、過剰歯の存在、永久歯の萌出障害といった問題に対しては、歯科的対応を行う。具体的には、補綴的対応、過剰歯の抜去と永久歯の外科的移動術、埋伏している永久歯を積極的に萌出させ配列するための外科的手法と矯正歯科的手法の組合せといったものがある。言語治療を必要に応じて行う。副鼻腔や中耳の感染症に対しても積極的治療を行い、反復性の中耳感染症に対しては、鼓膜切開・チューブ留置を検討し、インフルエンザ等の予防接種を定期的に行う。閉塞性睡眠時無呼吸の症候を示す例については、睡眠検査を行う。上気道閉塞に対しては外科的介入が必要になる場合がある。
定期的追跡評価 :
子どもに対しては、整形外科的合併症、歯の異常、洞・耳の感染症、上気道閉塞、難聴、スピーチの問題に関し、モニタリングを行う。骨密度の評価を目的としたDXAスキャンは、思春期初期から始め、その後は5年から10年間隔で行う。
避けるべき薬剤/環境:
高リスクの活動に参加するときは、ヘルメットや防具を着用する必要がある。
妊娠に関する管理:
罹患女性が妊娠したときは、児頭骨盤不均衡に関するモニタリングを行う。
遺伝カウンセリング
CCDスペクトラム障害は、常染色体顕性の遺伝形式をとる。CCDスペクトラム障害罹患者は、denovoの病的バリアントが大きな割合を占める。CCDスペクトラム障害罹患者の子にRUNX2の病的バリアントが継承される可能性は50%である。家系内に存在するRUNX2の病的バリアントが既知の場合には、出生前検査や着床前遺伝学的検査を行うことが可能である。
診断
鎖骨頭蓋異形成(CCD)スペクトラム障害は、古典的CCD(頭蓋縫合閉鎖遅延・鎖骨の低形成ないし無形成・歯の異常の3徴候)から、軽症型CCD、さらには骨格症候を伴わない歯の異常のみのものに至るまで、臨床的にもX線的にも1つの連続体を成す骨系統疾患である。CCDスペクトラム障害の正式な臨床診断基準は、今のところ確立していない。
本疾患を示唆する所見
以下のような臨床所見、X線所見を呈する発端者については、CCDスペクトラム障害を疑う必要がある。
臨床症候
- 出生時における異常に大きく開存した大泉門
大泉門の開存は、生涯にわたって続くことがある。
前頭縫合が開存することにより、前頭骨が正中の溝を隔てて左右に分離する。
額は幅が広く平坦で、頭蓋は短頭形を呈する。
- その他の頭蓋顔面症候
前額部や頭頂部の突出、眼間開離、中顔面の後退、低い鼻梁。
- 細いなで肩
鎖骨の低形成ないし無形成のため、両肩を正中で合わせられることがある(図1参照)。
- 歯の異常
第二生歯の萌出遅延、乳歯の脱落不全、さまざまな数の過剰歯とそれに伴う叢生、咬合異常。
- 指趾の異常
短指趾、先細りの指、幅広で短い拇指など。
- 低身長
低身長の程度は、通常、中等度。
- 正常の知能
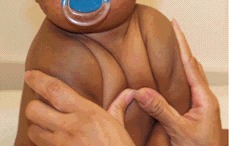
図1: 鎖骨の低形成を有する罹患者については、両肩を正中で合わせられることがある。
X線写真所見
- 頭蓋
- 大きく開いた縫合、大泉門の開存、ウォルム骨(縫合内の小骨)の存在
- 頭蓋骨の骨化遅延
- 副鼻腔、前頭洞、乳突洞の低含気ないし無含気
- 埋伏歯、叢生歯、過剰歯
- 胸郭
- 上部の直径が小さな円錐形の胸郭
- 通常、両側性(ただし、必ずしも影響の現れ方が対称的というわけではない)にみられる鎖骨の異常
これは、完全欠損から、低形成ないし鎖骨が不連続的なものまで、幅がみられる。
鎖骨への影響は、内側寄りに比べ、外側寄りのほうに顕著に現れる(図2参照)。
- 肩甲骨の低形成
- 骨盤
- 恥骨結合が大きく開いた恥骨の骨化遅延
- 腸骨翼の低形成
- 仙腸関節の拡大
- 大腿骨頸部が短く骨端が長くなった形の大腿骨頭の延長(シェフの帽子様外形)
- 内反股
- 手
- 中手骨・中足骨の偽骨端核
これにより第2中手骨の特徴的な延長がみられることがある(図3参照)。
- 末節骨の低形成
- 円錐形の骨端を伴う第3、第4、第5指中節骨の変形と短小化
- その他
DXAスキャンにて骨密度の低下が明らかになる骨減少症/骨粗鬆症。
中に、多発性骨折をきたす例がみられる。
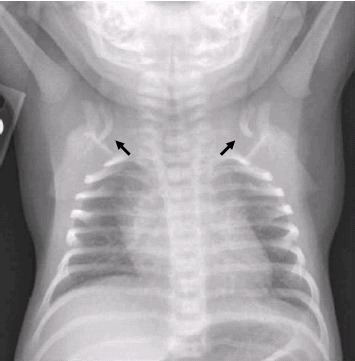
図2:鎖骨低形成を示す胸部X線写真
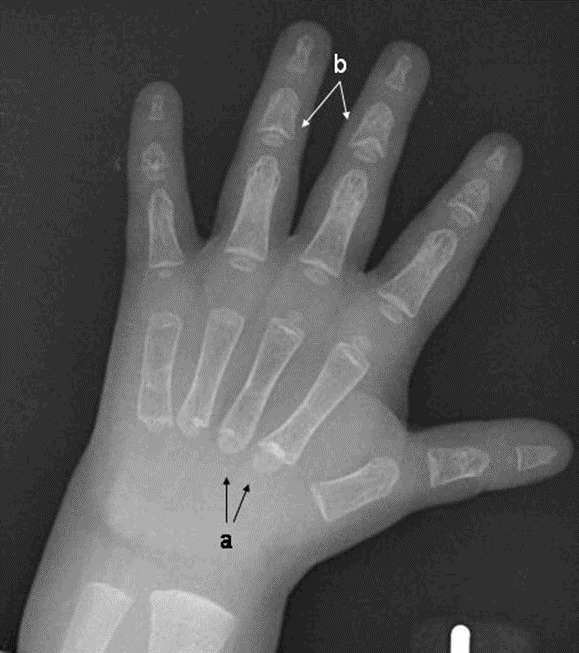
図3:鎖骨頭蓋異形成スペクトラム障害の2歳半男児の手のX線写真
a.第2、第3中手骨基部に偽骨端核がみられることに注目。
第4、第5中手骨基部には過剰成長軟骨板がみられる。
b.円錐形の骨端が、特に第3、第4中節骨に顕著にみられる。
指節骨の形成に異常があるように見受けられ、特に第2指から第5指の中節骨にそれが顕著にみられる。
診断の確定
発端者におけるCCDスペクトラム障害の臨床診断は、特徴的な臨床所見、X線写真所見がみられることで、また、発端者における分子診断は、これを示唆する所見がみられることに加え、分子遺伝学的検査でRUNX2にヘテロ接合性病的バリアント(pathogenicとlikelypathogenicの両方を含む)が同定されることをもって確定する(表1参照)。
注:(1)アメリカ臨床遺伝ゲノム学会(ACMG)/分子病理学会(AMP)のバリアントの解釈に関するガイドラインによると、「pathogenic」のバリアントと「likelypathogenic」のバリアントとは臨床の場では同義であり、ともに診断に供しうるものであると同時に、臨床的な意思決定に使用しうるものとされている[Richardsら2015]。本セクションで「病的バリアント」と言うとき、それは、あらゆるlikelypathogenicのバリアントまでを包含するものと理解されたい。
(2)ヘテロ接合性の意義不明バリアントが同定された場合、それは、本疾患の診断を確定するものでも否定するものでもない。
分子遺伝学的検査のアプローチとしては、単一遺伝子検査、マルチ遺伝子パネル、核型検査などが考えられる。
- 単一遺伝子検査
最初に、遺伝子内の小欠失/挿入、ナンセンス・スプライス部位バリアントを検出するためのRUNX2の配列解析を行う。
注:配列解析の手法によっては、単一エクソン、複数エクソン、遺伝子全体といったサイズの欠失/重複が検出されないことがある。そのため、配列解析でバリアントが検出されないようであれば、次いで、エクソン単位あるいは遺伝子全体の欠失や重複を調べるためのRUNX2の遺伝子標的型欠失/重複解析を行うようにする。
- マルチ遺伝子パネル
現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝学的原因を特定する上では、RUNX2その他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネルも検討に値する。
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
- 核型検査
CCDスペクトラム障害の症候を示すと同時に、他の先天異常症候群や発達遅滞を併せもつ例で、RUNX2の検査で診断に至らなかった場合には、6p21.1領域(RUNX2の座位)が関与しつつもRUNX2のコピー数変化には至らない複雑な染色体再構成や転座を調べるための核型検査も検討対象になろう。
表1:鎖骨頭蓋異形成スペクトラム障害で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的バリアント2が検出される発端者の割合 |
|---|---|---|
| RUNX2 | 配列解析3 | 70%-80%近く4 |
| 遺伝子標的型欠失/重複解析5 | 15%近く4,6 | |
| 核型検査 | 脚注7参照 | |
| 不明 | 適用対象外 | 5%-15%近く |
- 染色体上の座位ならびにタンパク質に関しては、表A「鎖骨頭蓋異形成スペクトラム障害:遺伝子とデータベース」を参照。
- 本遺伝子において検出されている各種バリアントに関する情報については、「分子遺伝学」の項を参照。
- 配列解析を行うことで、benign、likelybenign、意義不明、likelypathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位ないし遺伝子全体の欠失や重複については検出されない。配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
- Ottら[2010]、Motaeiら[2021]、ヒト遺伝子変異データベース(HGMD)の購読ベースの専門家向けデータ[Stensonら2020]、ならびにClinVarデータベースのレビューに基づく。
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失ないし重複の検出を目的に設計された遺伝子標的型マイクロアレイなどがある。遺伝子標的型欠失/重複解析では、単一エクソンから遺伝子全体といったサイズの欠失が検出されるが、大きな欠失の切断点、隣接遺伝子(例えば、Puvabanditsinら[2018]の報告にあるもの)の切断点などは、こうした手法では特定できない可能性がある。
- RUNX2の欠失を有する罹患者は、CCDスペクトラム障害の表現型に加えて、発達遅滞などの別の所見も示す場合がある。
- RUNX2の座位を含む転座を示した2例が報告されている[Purandareら2008,Northupら2011]。
臨床的特徴
臨床像
鎖骨頭蓋異形成(CCD)スペクトラム障害は、古典的CCD(頭蓋縫合閉鎖遅延・鎖骨の低形成ないし無形成・歯の異常の3徴候)から、軽度型CCD、さらには骨格症候を伴わない歯の異常のみのものに至るまで、臨床的連続性を示す骨系統疾患である[Golanら2000]。
古典的症候を有していることで診断に至る例が大多数を占める。CCDスペクトラム障害では、頭蓋や鎖骨のような膜性骨化を示す骨に最も強い影響が及ぶものの、軟骨内骨化により形成される骨についても、影響を受けることがある。Cooperら[2001]は、90人の発端者、ならびに56人の第1度・第2度近親について、自然歴を報告しており、その中で、同一の病的バリアントを共有する同一家系内の罹患者間にあってさえ、臨床的表現型には大きなばらつきがあることを強調している。Robertsら[2013]は、南アフリカで自らの経験した100人以上の罹患者についてレビューを行っている。
古典的CCD
古典的CCD罹患者で最も顕著にみられる所見は、「本症候群を示唆する所見」で述べた通り、出生時において異常に大きく開き、生涯にわたって開存する可能性のある大泉門、鎖骨の低形成によって生じる正中で左右の肩を合わせることのできる細いなで肩、歯の異常(本セクション中の「歯科的合併症」の項を参照)である。
CCDスペクトラム障害罹患者においてみられるその他の医学的問題としては、低身長、骨格的/整形外科的所見、歯科的合併症、耳鼻咽喉科的合併症、内分泌所見、軽度の発達遅滞といったものがある。
身長
CCDスペクトラム障害罹患者の多くは非罹患同胞より低身長で、出生後の発育不全を示す。
- 男性罹患者は、非罹患者の男性同胞に比べ平均で6インチ低く、平均身長は165cm(±8cm)である。
- 女性罹患者は、非罹患者の女性同胞に比べ平均で3インチ低く、平均身長は156cm(±10cm)である[Cooperら2001]。
骨格的/整形外科的所見
罹患者は、骨に関連するその他の諸問題を有することが多い。
- 扁平足(57%)
- 外反膝(X脚)(28%)
- 脊柱側彎(18%)[Cooperら2001]
- 骨粗鬆症がCCDスペクトラム障害罹患者14人中8人(57%)、骨減少症が14人中3人(21%)で確認されている[DinçsoyBirら2017]。
これらより低頻度の整形外科的問題としては、肩関節や肘関節の脱臼がある[El-Gharbawyら2010]。
歯科的合併症
CCDスペクトラム障害罹患者の94%に、第二生歯萌出障害と乳歯脱落不全をはじめとする歯科的問題がみられる[Golanら2003]。CCDスペクトラム障害罹患者に最も共通した歯科的所見は、第二大臼歯と乳歯の混在(80%)、下顎切歯部の大きな空隙、過剰歯胚の存在(70%)、左右下顎枝の平行化である[Cooperら2001,Golanら2003,Golanら2004,Bufalinoら2012]。CCDスペクトラム障害罹患者は下顎前突になりがちで、顎骨内の過剰歯周囲に嚢胞を形成しがちである[McNamaraら1999]。
耳鼻咽喉科的合併症
一般集団に比べ、CCDスペクトラム障害罹患者においては、反復性の洞感染症その他の上気道の合併症が明らかに生じやすい傾向にある。上気道閉塞を示唆する症候がみられるときは、睡眠検査が適応となり、場合によっては外科的介入を要するようなこともある。罹患者の39%に伝音性難聴がみられる。年齢を問わず、CCDスペクトラム障害罹患者には反復性の耳の感染症が多くみられる。
内分泌
CCDスペクトラム障害罹患者の中に、IGF-1レベルが低値を示す例がみられる。これまでに、骨粗鬆症という形での直接的影響が現れていないビタミンD低下の報告もみられる[DinçsoyBirら2017]。CCDスペクトラム障害罹患者は、稀にアルカリホスファターゼ値の低下を示すことがある[Moravaら2002,Ungerら2002,El-Gharbawyら2010]。
発達
通常、知能は正常である。5歳未満の子どもについては、軽度の運動発達遅滞、特に粗大運動技能の遅延を示すことがある。扁平足や外反膝などの整形外科的合併症に起因して、こうした遅延が生じる場合がある。小学生になると、周りの子どもたちに比べ、明らかな違いはみられなくなる。
遺伝型-表現型相関
CCDスペクトラム障害でみられる歯の症候については、遺伝型と表現型との間の相関が一部明らかになっている。遺伝型と鎖骨の病変との間の相関については、明確な形にはなっていない[Ottoら2002,Bufalinoら2012,Jarugaら2016]。
- RUNX2の、runtドメイン内に位置する病的バリアント(もしくはruntドメイン内やその上流で未成熟終止をきたすことが予測される病的バリアント)のヘテロ接合により、変異タンパク質のトランス活性化活性の消失を招き、その結果生じるハプロ不全により古典的CCDが発生する。
- 古典的CCDの表現型を示す例について言うと、runtドメインに影響を及ぼす病的バリアントを有する罹患者に比べ、runtドメインは正常でRUNX2活性が高いレベルで残っている罹患者のほうが、低身長や歯の異常の程度が軽度にとどまることが確かめられている[Yoshidaら2002]。
- 骨格症候を伴わない歯の異常単独のCCDから、軽度のCCD、さらには古典的CCDまで、幅広い臨床スペクトラムをもたらすものとして、タンパク質機能の部分的喪失をきたすハイポモルフィックな病的バリアント(c.1171C>T[p.Arg391Ter],c.598A>G[p.Thr200Ala],c.90dupC[p.Ser31LeufsTer130])(「分子遺伝学」の項を参照)がある。
その場合の家系内のばらつきの幅はかなり大きい[Zhouら1999]。
- 反復性骨折や脊柱側彎をきたすような骨粗鬆症は、病的フレームシフトバリアントc.1205dupCのヘテロ接合に起因して生じる。これは、成人の骨を維持していく上でのRUNX2タンパク質の役割を反映したものである[Quackら1999]。
浸透率
RUNX2の病的バリアントは高い浸透率を示す。今のところ、不完全浸透を示唆する報告はみられない。
疾患名について
鎖骨頭蓋異形成スペクトラム障害は、大家系中の数人にみられた歯-骨異形成症として最初に報告されたものである。
本疾患に対しては、かつては「鎖骨頭蓋異骨症」という用語が用いられたものの、RUNX2が骨の形成や維持に重要な役割を果たしていることを踏まえ、現在では異形成症という、より正確な捉えられ方がなされている。
発生頻度
Stevensonら[2012]は、アメリカ、ユタ州の集団について、10,000人あたり0.12人という数字を示し、それまで認識されていたより発生頻度が高い可能性を示唆している。
遺伝学的に関連のある疾患(同一アレル疾患)
RUNX2の遺伝子内部分重複により、骨幹端異形成-上顎低形成-短指症(metaphysealdysplasia,maxillaryhypoplasia,andbrachydactyly;MDMHB)(訳注:「2019年版骨系統疾患国際分類の和訳」では、「上顎低形成を伴う骨幹端異形成症」と訳されている)(OMIM156510)が生じる。
罹患者には、低身長、長管骨と脊椎の異常、歯のジストロフィー、鎖骨内側半分の拡大がみられる。
頭蓋縫合早期癒合症と部分性無歯症を呈する複数の罹患者で、RUNX2の完全重複が報告されている[Meffordら2010,Greivesら2013,Molinら2015]。
鑑別診断
鎖骨頭蓋異形成(CCD)スペクトラム障害と一部の症候を共有する疾患がいくつか存在する。CCDスペクトラム障害と共通の骨格要素が影響を受けることから考えて、こうした疾患は、下流ターゲットに対するRUNX2の作用に影響を及ぼす遺伝子群(うち、最も注目すべきはCBFB)の変異に起因するものであるように思われる(CBFBは、RUNX2とヘテロ二量体を形成して下流のターゲットの転写を活性化する)。
表2:CCDスペクトラム障害との鑑別診断に関連してくる遺伝子群
| 遺伝子/遺伝学的メカニズム | 疾患名 | 遺伝形式 | 頭蓋顔面と歯の症候 | 骨格の症候 | その他の症候 |
|---|---|---|---|---|---|
| ALPL | 低ホスファターゼ症1 | AR AD2 |
|
|
|
| CBFB(遺伝子内病的バリアント) | CBFB関連鎖骨頭蓋異形成症(OMIM620099) | AD |
|
|
|
| CBFB(CBFBを含む16q22.1の欠失) | 16q22欠失症候群(OMIM614541) | AD | 大きく開いた大泉門 |
|
|
| CTSK | 濃化異骨症 | AR | 大泉門の開存を伴う頭蓋縫合の閉鎖不全 |
|
|
| FIG4 | YunisVaron症候群(OMIM216340) | AR |
|
|
|
| LMNA ZMPSTE24 |
下顎先端症候群(OMIMPS248370) | AR |
|
|
|
| MSX2(遺伝子内病的バリアント) | 鎖骨頭蓋異形成症を伴う頭頂孔4 | AD |
|
鎖骨低形成 | |
| MSX2(MSX2の上流の微小重複) | MSX2関連多合指趾を伴う鎖骨頭蓋異形成症5 | AD | 鎖骨頭蓋異形成症の表現型模写 | 鎖骨頭蓋異形成症の表現型模写 | 多合指趾が一部の例でみられる |
AD=常染色体顕性,AR=常染色体潜性
- 重度のCCD罹患者で、当初、低ホスファターゼ症とみられていた1例の報告がある[Ungerら2002]。
- 周産期型ならびに乳児型の低ホスファターゼ症は、通常、常染色体潜性の遺伝形式をとる。より軽度型のもの、特に成人型、歯限局型低ホスファターゼ症については、ALPLの病的バリアントが組織非特異型アルカリホスファターゼ(TNSALP)の活性に及ぼす影響に従って、常染色体潜性もしくは常染色体顕性の遺伝形式をとる。
- Gotoら[2004]
- 「頭頂孔拡大」のGeneReviewを参照。
- Ottら[2012]
CCDスペクトラム障害の鑑別診断に関係してくるその他の疾患/状況
- Crane-Heise症候群(OMIM218090)
これは致死性の疾患で、遺伝学的原因は明らかになっていない。
頭蓋冠の石灰化不良、大頭症、口唇裂口蓋裂、低位で異形成の耳介、頸椎の欠損を伴う重度の脊椎・四肢奇形、鎖骨・肩甲骨の低形成、指節骨の低形成と欠損、多発性関節拘縮、子宮内発育不全、性器低形成などがみられる。
- CDAGS症候群(OMIM603116)
RNU12をコードする核内低分子RNAの両アレル性病的バリアントに起因して生じる疾患で[Xingら2021]、縫合閉鎖の促進・骨化遅延という、見かけ上相反する病態生理学的プロセス、発生プロセスの組合せがみられる[Mendoza-Londonoら2005]。
CDAGS症候群では、大泉門の閉鎖遅延、鎖骨低形成、頭蓋縫合早期癒合症、肛門奇形、尿路性器奇形、皮膚病変(汗孔角化症)がみられる。
- 家族性過剰歯
非症候群性の小臼歯部過剰歯[Baeら2017]。
- 甲状腺機能低下症
甲状腺機能低下症においても大泉門の閉鎖遅延がみられることがある。
臨床的マネジメント
鎖骨頭蓋異形成(CCD)スペクトラム障害に関する臨床的管理のガイドラインは、今のところ公表されていない。
最初の診断に続いて行う評価
CCDスペクトラム障害と診断された罹患者については、疾患の範囲やニーズを把握するため、診断に至る過程ですでに実施済でなければ、表3にまとめた評価を行うことが推奨される。
表3:鎖骨頭蓋異形成スペクトラム障害罹患者の最初の診断後に行うことが推奨される評価
| 系/懸念事項 | 評価 | コメント |
|---|---|---|
| 骨格 |
|
|
| 二重エネルギーX線吸収測定法(DXAスキャン) | 骨減少症のリスクがあるため、思春期初期にDXAスキャンを行う必要あり。 | |
| 歯 | CCDスペクトラム障害とその管理に精通した歯科医による歯科的評価 | |
| 聴覚 | 聴覚評価 | |
| 遺伝カウンセリング | 遺伝の専門医療職1の手で行う。 | 医学的、個人的な意思決定の用に資するべく、本人や家族に対し、CCDスペクトラム障害の本質、遺伝形式、そのもつ意味についての情報提供を行う。 |
- 臨床遺伝医、認定遺伝カウンセラー、認定上級遺伝看護師をいう。
症候に対する治療
生活の質の改善、機能の最大限の賦活、合併症の低減といったことを目的とした支持療法が推奨される。関連各分野の専門家による多職種連携管理とするのが理想的である(表4参照)。
表4:鎖骨頭蓋異形成スペクトラム障害罹患者の症候に対する治療
| 症候/懸念事項 | 治療 | 考慮事項/その他 |
|---|---|---|
| 頭蓋顔面症候 |
|
大多数の例で、大泉門は経時的に閉鎖するため、通常は、頭蓋の形態修正術を要しない。 |
| 麻酔に伴う気道管理 |
|
歯や頭蓋顔面に異常があると、気道管理がそれだけ難しくなると予測される。 |
| 鎖骨低形成 | 一部の例については、美容的観点から、低形成の鎖骨に対する延長術の検討がなされる1。 | |
| 骨粗鬆症 | DXAスキャンで骨密度が正常範囲を下回る例については、カルシウムとビタミンDの補充を行う。 | |
| 歯の症候 | 必要な処置を適切な時期に計画できるようCCDに精通した歯科医への紹介を早期に行う。 | 一般的には、乳歯を抜去して永久歯の開窓を行う複数の口腔外科的手術を協調させながら積極的に行っていくやり方が推奨される。 最初の萌出遅延が生じた後は、自然萌出を期待して注意深く経過観察を続けるというやり方は有効ではない3。 |
治療を要する歯科的症候:
|
||
治療の目標:
|
||
目標を達成するための手段:
|
||
| スピーチの問題 | 歯科治療を行っている期間や、聴覚障害に起因するスピーチの問題を抱えている例については、必要に応じ言語治療を行う。 | |
| 洞や中耳の感染4 |
|
|
| 上気道閉塞 |
|
|
| 低身長 | 治療は推奨されない。 |
|
- ごく少数例の報告ながら、前頭縫合部の陥凹と鎖骨低形成に対する外科的介入が成果を収めたとの報告がみられる[Kangら2009,Sewellら2013]。
- Ioscovichら[2010]
- 詳細なレビューについては、Beckerら[1997a]、Beckerら[1997b]、Robertsら[2013]、Farrowら[2018]を参照されたい。
- Visoskyら[2003]を参照。
- Zhengら[2005]
定期的追跡評価
現状の症候のモニタリング、支持療法に対する罹患者の反応様相のモニタリング、新たな症候の出現様相のモニタリングといったことを目的として、表5にまとめたような評価を行うことが推奨される。
表5:鎖骨頭蓋異形成スペクトラム障害罹患者で推奨される定期的追跡評価
| 系/懸念事項 | 評価 | 実施頻度 |
|---|---|---|
| 骨格 | 骨格症候に関する整形外科的評価(扁平足,外反膝,脊柱側弯,反復性肩関節・肘関節脱臼) | 小児期を通じ、来院ごと |
| 骨密度測定のためのDXAスキャン(骨粗鬆症の評価のため) | 思春期初期(あるいは、骨折の増加をはじめとする骨減少症の症候を呈する例についてはさらに早く)から始め、5-10年に1度 | |
| 歯 | CCDスペクトラム障害とその管理に精通した歯科医による歯科的評価 | 3歳から始め、6ヵ月に1度、もしくは歯科医の推奨に従ってさらに高頻度に |
| 耳鼻咽喉 |
|
来院ごと |
| 聴覚評価 | 反復性の耳感染症を有する例については年に1度 | |
| 発達 | 言語評価 | 歯科治療中、ならびに反復性感染症や聴力の問題を有する例について、来院ごと |
避けるべき薬剤/環境
頭部の外傷を回避するため、高リスクのスポーツや活動に参加する際には、ヘルメットや防具を着用すべきである。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
妊娠に関する管理
CCDスペクトラム障害の妊婦については、児頭骨盤不均衡に関するモニタリングを慎重に行う必要がある。児頭骨盤不均衡がある場合は、帝王切開による分娩が必要になる可能性がある。CCDスペクトラム障害女性の初産の際の帝王切開施行率は69%と、対照群より高い値を示す[Cooperら2001]。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「ClinicalTrials.gov」、ならびにヨーロッパの「EUClinicalTrialsRegister」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
鎖骨頭蓋異形成(CCD)スペクトラム障害は、常染色体顕性の遺伝形式をとる。
家族構成員のリスク
発端者の両親
- CCDスペクトラム障害と診断された罹患者の一部は、罹患者である片親からの継承例である。
- RUNX2に生じたdenovoの病的バリアントのヘテロ接合に起因してCCDスペクトラム障害が生じる例もある。罹患者に占めるdenovoの病的バリアントの比率は高い。
- 発端者が家系内で唯一の罹患者(すなわち、孤発例)と目される場合に、発端者の両親に対して行うことが推奨される評価は以下の通りである。
- 歯や骨の異常をうかがわせる徴候がみられる場合は、詳しい臨床診査、ならびに頭蓋顔面や骨格のX線写真撮影を検討する。
(注:同一の病的バリアントを共有する親子間にあっても、表現型が大きく異なるということがありうる。)
- 発端者の分子診断が確定している場合には、両親の遺伝学的状態を確認して再発リスクに関するカウンセリングの信頼性を担保することを目的として、両親に対し分子遺伝学的検査を行う。
- 発端者で同定された病的バリアントが両親いずれからも検出されず、なおかつ、親子鑑定の結果、両者が真正の生物学的父・母であることが確認されている場合には、可能性として次のことを検討する必要がある。
- 発端者に生じたdenovoの病的バリアントである可能性。
- 生殖細胞系列モザイク(もしくは、体細胞・生殖細胞系列両方のモザイク)の片親から、発端者が病的バリアントを継承した可能性*[Palら2007,Qianら2018,Muurinenら2022]。
*RUNX2の病的バリアントを体細胞・生殖細胞系列の両方で有する片親は、軽度、ないしごく軽微な症候を呈することが考えられる。
- CCDスペクトラム障害と診断された罹患者の中には、家系内のメンバーが本疾患を有していることを見逃してしまったために、見かけ上、家族歴陰性であるかのように認識されてしまう例もみられる。したがって、両親に対して適切な臨床的診査(骨格のX線写真撮影も含めて)を行うか、分子遺伝学的検査(発端者で同定された病的バリアントに関し、いずれの親もヘテロ接合体ではないことを確認する検査)を行うといったことをしない限り、家族歴陰性の確定はできない。
発端者の同胞
-
発端者の同胞の有するリスクは、発端者の両親の遺伝学的状態によって変わってくる。
- 発端者の片親も罹患者であった、ないしは、発端者で同定された病的バリアントを片親も有していたといった場合であれば、同胞の有するリスクは50%である。
- RUNX2の同一の病的バリアントを継承した同胞間で、現れる表現型に相違がみられるということがありうる(RUNX2の病的バリアントは高い浸透率を示すものの、臨床症候の現れ方については、家系内の罹患者間でかなり大きなばらつきの幅がみられる;「遺伝型-表現型相関」ならびに「浸透率」の項を参照)。
- 発端者で同定されたRUNX2の病的バリアントが、両親いずれの白血球DNAからも検出されない場合、同胞への再発リスクは、一般集団より若干高い程度と推定される。それは、片親が生殖細胞系列モザイクであるという可能性が残るからである。見かけ上、非罹患者の母親から3人の罹患同胞が生まれた1家系で、生殖細胞系列モザイクが確認された例が実際に存在する[Palら2007]。
- 臨床的にみて両親とも非罹患者と思われるものの、その遺伝学的状態までは明らかになっていないといった場合の発端者の同胞の有するリスクは、低いとは思われるものの、それでも一般集団よりは高くなる。それは、片親が生殖細胞系列モザイクであるという可能性が残るからである。
発端者の子
CCDスペクトラム障害罹患者の子にRUNX2の病的バリアントが継承される確率は50%である。
他の家族構成員
他の血族の有するリスクは、発端者の両親の状態によって変わってくる。片親にCCDスペクトラム障害の症候がみられる、あるいは片親がRUNX2の病的バリアントを有しているということになれば、その血族に当たる人もリスクを有することになる。
関連する遺伝カウンセリング上の諸事項
家族計画
- 遺伝的リスクの確定、出生前/着床前遺伝学的検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者である若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原メカニズム・疾患等に対するわれわれの理解は、将来、より進歩していくことが予想される。そのため、分子診断が未確定の(すなわち、原因となった病原メカニズムが未解明の)発端者のDNAについては、保存しておくことを検討すべきである。より詳細な情報については、Huangら[2022]を参照されたい。
出生前検査ならびに着床前遺伝学的検査
分子遺伝学的検査
家系内に存在するRUNX2の病的バリアントの内容が確定している場合には、CCDスペクトラム障害に関する出生前検査や着床前遺伝学的検査を行うことが可能である。
超音波検査
親が罹患者である場合、古典的CCDに関しては、妊娠14週になれば、超音波検査によって子の診断を行うことが可能となる。一貫してみられる症候として最も多いのは鎖骨の異常で、短小(妊娠週齢で比較したとき5パーセンタイル未満)、部分欠損、全欠損などがみられる。これより出現頻度の低い特異的所見として、他に、低石灰化を伴う短頭形の頭蓋、前額部の突出、全身性の骨化遅延などがある[Stewartら2000,Hermannら2009]。
注:妊娠週齢については、最終月経の第1日から数えた週齢、もしくは超音波での計測値をもとにした週齢で表記される。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- AboutKidsHealth
- CCDSmiles
- Children'sCraniofacialAssociation
- FACES:NationalCraniofacialAssociation
- HumanGrowthFoundation
- MAGICFoundation
- UCLAInternationalSkeletalDysplasiaRegistry(ISDR)
Canada
Cleidocranial Dysplasia (CCD)
Phone:800-535-3643
Email:contactCCA@ccakids.com
www.ccakids.org
Phone:800-332-2373;423-266-1632
Email:info@faces-cranio.org
www.faces-cranio.org
Phone:800-362-4423;630-836-8200
Fax:630-836-8181
Email:contactus@magicfoundation.org
www.magicfoundation.org
Phone:310-825-8998
International Skeletal Dysplasia Registry
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:鎖骨頭蓋異形成スペクトラム障害:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specific データベース |
HGMD | ClinVar |
|---|---|---|---|---|---|
| RUNX2 | 6p21.1 | RRunt関連転写因子2 | RUNX2 database | RUNX2 | RUNX2 |
データは、以下の標準資料から作成したものである。遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:鎖骨頭蓋異形成スペクトラム障害関連のOMIMエントリー(内容の閲覧はOMIMへ)
| 119600 | CLEIDOCRANIAL DYSPLASIA 1; CLCD1 |
| 600211 | RUNT-RELATED TRANSCRIPTION FACTOR 2; RUNX2 |
分子レベルの病原
RUNX2は、runt関連転写因子2(RUNX2)をコードする。RUNX2は、骨芽細胞の分化や骨格の形態形成に関与する転写因子である。RUNX2は、膜性骨化の際の骨芽細胞の分化、ならびに軟骨内骨化の際の軟骨細胞の成熟に不可欠なものである[Zhengら2005]。RUNX2は、N末端にQ/Aドメインと呼ばれるポリグルタミンとポリアラニンの反復構造をもつアミノ酸残基ストレッチ、1つのruntドメイン、C末端にプロリン/セリン/スレオニン(PST)リッチ活性化ドメインを有する。Runtドメインは、もともとショウジョウバエのrunt遺伝子で発見された128個のアミノ酸から成るポリペプチドモチーフで、DNAとの結合やタンパク質の二量体形成に関し、独立メディエーターとしての独特の働きをしている[Zhouら1999]。
古典的CCD罹患者で確認されているRUNX2の病的バリアントは、その大部分がruntドメインに影響を及ぼすもので、病的バリアントの大半はDNAとの結合能力の喪失につながるものと考えられている[Leeら1997,Mundlosら1997,Ottoら2002]。RUNX2の機能上、アルギニン225(p.Arg225)は決定的に重要な残基であるが、病的ミスセンスバリアントはこの部分に集中している。Invitroの研究で、p.Arg225の病的ミスセンスバリアントによりRUNX2タンパク質の核内蓄積が阻害されることがわかっている。
タンパク質の部分的機能喪失をきたすRUNX2のハイポモルフィックなアレルであるc.90dupCとc.598A>Gにおいては、軽度型のCCD、歯の異常のみのCCD、家系内で大きな表現型のばらつきを示すタイプのCCDといったものを生じる。
疾患の発症メカニズム
機能喪失型である。
RUNX2特異的な検査技術上の考慮事項
ゲノムのレベルで言うと、RUNX2の最長の転写産物バリアント(NM_001024630.4)には9つのエクソンがある。別のタンパク質アイソフォームをコードする転写産物バリアント[Geoffroyら1998]は、代替プロモーターを用いた選択的スプライシングにより産生される。
表6:RUNX2の注目すべき病的バリアント
| 参照配列 | DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | コメント |
|---|---|---|---|
| NM_001024630.4 NP_00109801.3 |
c.90dupC | p.Ser31LeufsTer130 | ハイポモルフィックアレル(「遺伝型-表現型相関」の項を参照) |
| c.598A>G | p.Thr200Ala | ||
| c.673C>T | p.Arg225Trp | 「分子レベルの病原」の項を参照。 | |
| c.674G>T | p.Arg225Leu | ||
| c.674G>A | p.Arg225Gln | ||
| c.1171C>T | p.Arg391Ter | ハイポモルフィックアレル(「遺伝型-表現型相関」の項を参照) | |
| c.1205dupC | p.Pro403AlafsTer871 | 骨粗鬆症から反復性骨折に至る[Quackら1999] |
上記のバリアントは報告者の記載をそのまま載せたもので、GeneReviewsのスタッフが独自に変異の分類を検証したものではない。
GeneReviewsは、HumanGenomeVariationSociety(varnomen.hgvs.org)の標準命名規則に準拠している。
命名規則の説明については、QuickReferenceを参照のこと。
- コドンPro402のフレームシフト変異として報告されたもの[Quackら1999]
更新履歴:
-
Gene Reviews著者: Karen Machol, MD, PhD,Roberto Mendoza-Londono, MD, MS, Brendan Lee MD, PhD
日本語訳者:佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学) - GeneReviews最終更新日: 2017.11.16 日本語訳最終更新日: 2023.1.8
Gene Reviews著者: KarenMachol,MD,PhD,RobertoMendoza-Londono,MD,MS,andBrendanLeeMD,PhD
日本語訳者: 佐藤康守(たい矯正歯科)、森貞直哉(兵庫県立こども病院)
GeneReviews最終更新日: 2023.4.13. 日本語訳最終更新日: 2023.8.6.[in present]
原文: Cleidocranial Dysplasia Spectrum Disorder
![]()