CHD7疾患
(CHD7 disorder)
Gene Reviews著者: Conny M van Ravenswaaij-Arts, MD, PhD, Meg Hefner, MS, Kim Blake, MD, MRCP, FRCPC, and Donna M Martin, MD, PhD
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.9.29. 日本語訳最終更新日: 2022.10.1.
要約
疾患の特徴
CHD7疾患というのは、CHD7の病的バリアントのヘテロ接合が形作るスペクトラム全体を包含する疾患名である。
そこには、CHARGE症候群だけでなく、CHARGE症候群の表現型を構成するサブセットとしての症候がすべて含まれる。
覚えやすさを意識したCHARGE症候群という名前は、分子レベルになる前の時代の命名で、コロボーマ(coloboma)、心奇形(heart defect)、後鼻孔閉鎖(choanal atresia)、成長発達遅滞(retarded growth and development)、生殖器系の低形成(genital hypoplasia)、耳の奇形(ear anomaly;難聴を含む)を表している。
CHD7疾患は、その後、遺伝的原因が特定されるに至り、続いてその表現型のスペクトラムが、脳神経奇形、前庭奇形、口唇裂口蓋裂、甲状腺機能低下、気管食道奇形、脳奇形、癲癇、腎奇形を包含するところにまで拡大していった。
寿命は、症候の重症度によって大きく変わってくる。
出生時に重度の奇形(特に複雑心奇形)があって、さらに気道や摂食の障害によって問題が複雑化していることも多く、そうした場合は、生後数年のうちに死亡する率も高い。
小児期、思春期、成人期における死亡事例は、残存している心奇形の状態、感染、誤嚥ないし窒息、閉塞性・中枢性無呼吸を含む呼吸の問題、そしてことによると癲癇なども含め、さまざまな要因が組み合わさって生じるものであるように思われる。
診断・検査
発端者におけるCHD7疾患の診断は、本疾患を示唆する臨床所見や画像所見がみられることに加えて、分子遺伝学的検査でCHD7の病的バリアントのヘテロ接合、ないしCHD7の欠失が確認されることで確定する。
臨床的マネジメント
症状に対する治療:
CHD7疾患の症候に対する管理は複雑になることがあり、医師、各分野の専門療法士、教育者を含む集学的アプローチが必要になるような場合もある。
定期的追跡評価:
乳児期から小児期にかけて確認された症候を定期的にフォローすること、ならびに、成長、発達、教育の進行状況、行動、そして場合によっては内分泌の問題なども含めて、継続的にモニターしていくことが必要である。
避けるべき薬剤/環境:
全身麻酔後、気道の合併症のリスクが高まるため、全身麻酔を必要とするような処置は最小限の回数に抑え、可能な限り複数の処置を1回で行うよう努める。
遺伝カウンセリング
CHD7疾患は常染色体顕性遺伝疾患で、その多くが新生の病的バリアントによって引き起こされる。
稀に、CHD7の病的バリアントのヘテロ接合を有する親からの継承例もみられる。
発端者の同胞への発症リスクは、親の遺伝的状態によって変わってくる。
(1)発端者の片親がCHD7の病的バリアントを有している場合、発端者の同胞がその病的バリアントを継承するリスクは50%である。
(2)発端者で同定されたCHD7の病的バリアントがいずれの親の白血球DNAからも検出されなかった場合、発端者の同胞への再現リスクは、経験的に言っておおむね1-2%といったところである。
それは、親の生殖細胞モザイクの可能性が残っているからである。
CHD7疾患罹患者の多くは生殖能力を有しないものの、子をなした場合、子がCHD7の病的バリアントを継承する確率は50%である。
家系内に存在するCHD7の病的バリアントが同定されている場合には、出生前、着床前遺伝子検査を行うことが可能である。
GeneReviewの視点
現在は、マルチ遺伝子パネル検査や網羅的ゲノム検査が広く行われるようになっている。
それに伴って明らかになってきたことは、CHD7の病的バリアントのヘテロ接合が示す表現型のスペクトラムは、単なるCHARGE症候群の範囲を超えて、CHARGE症候群の表現型を構成するサブセットとしての症候を広く包含するところにまで広がるということである。
本章のタイトルである「CHD7疾患」という言葉の意味するところは、これがCHD7の病的バリアントのヘテロ接合に関連して生じる表現型のスペクトラム全体を指す疾患名であるという意味合いが1つ、そしてそれと同時にもう1つ、このバリアントが同定された罹患者に対しては、表現型のスペクトラム全体に関して(分子遺伝学的検査に進むきっかけとなった臨床症候が何であったかとは無関係に)、可能な限りの医学的対応を行う必要性、ならびに、罹患者家族へのカウンセリングに際して、CHD7の病的バリアントが発見されたということのもつ意味が、CHARGE症候群という診断名と同一のものではないと説明することの重要性を強調するところにある。
診断
本疾患を示唆する所見
以下のような所見や家族歴の組合せを有する例に対しては、CHD7疾患を疑う必要がある。
臨床所見ならびに画像所見
- 虹彩、網膜、脈絡膜、視神経乳頭のコロボーマ、ないし無眼球や小眼球。
- 後鼻孔の閉鎖ないし狭窄:これは片側性のこともあれば両側性のこともある。
- そして、骨性のこともあれば粘膜性のこともあり、これについては非造影の軸位CTの体軸断面で確認可能である。
- 口蓋裂もしくは口唇口蓋裂(注:口蓋裂を伴う例については、後鼻孔閉鎖はほとんどみられない)
- 脳神経の機能障害ないし奇形
- 第Ⅰ脳神経:嗅覚脱失ないし嗅覚鈍麻
- 第Ⅶ脳神経:顔面神経麻痺(片側性ないし両側性)
- 第Ⅷ脳神経:感音性難聴ないし平衡覚障害、画像上で確認される第Ⅷ脳神経の低形成ないし無形成
- 第Ⅸ,第Ⅹ脳神経:吸啜/嚥下障害ならびに誤嚥、腸管運動障害
- 耳の奇形(CHD7疾患ではこれが最も特徴的)
- 耳介:短く幅広で、耳垂はほとんどあるいは全くない。
切り取られたような耳輪、対珠(訳注:原文では「耳珠」となっているが、図1の説明文では「対珠」となっている。「対珠」が正しいように思われる)とはつながらないことが多い突出した対耳輪、三角形の耳甲介、軟骨減少、しばしば突出し、多く非対称な耳介(図1参照)
- 中耳:耳小骨奇形(混合性難聴のため、オージオグラムは典型的な楔型を示す)
- 側頭骨の異常(多くの場合、側頭骨のCTスキャンで確認される):蝸牛のMondini型奇形、半規管の欠損ないし低形成
- 気管食道瘻ないし食道閉鎖
- 円錐動脈幹奇形(例えばFallot四徴)、共通房室弁口、大動脈弓異常などの心血管系奇形[Corsten-Janssen & Scamber 2017]
- 低ゴナドトロピン性性腺機能低下
- 出生時の男性:小陰茎と停留精巣
- 出生時の女性:陰唇低形成,子宮の異常ないし(稀ではあるが)欠損
- 男女とも:思春期が遅延ないし欠落し、しばしば嗅覚脱失を伴う[Bergmanら2011a]
- 発達遅滞/知的障害、ならびに運動発達指標の遅れ
後者は、感覚障害・平衡覚障害に伴う二次性のものであることが多い。
- 成長障害:通常、出生後に生じる低身長
成長ホルモン分泌不全を伴うことも伴わないこともある。
- 他の臨床症候
- 顔:四角い顔
- 広い額、広い鼻梁、目立つ鼻柱、平坦な頰骨部、顔面神経麻痺その他に伴う非対称、口唇裂、小さなオトガイ(これは年齢とともに大きく広くなる)(図2参照)
- 頸:頸は短く幅広で、なで肩[O‘Gradyら2016](図2参照)
- 手:通常、手掌は短く広い。
ホッケースティック形の屈曲線、短指、第2-5指の形に似た拇指(図3参照)を伴う。
割合は小さいながら、多指、欠指もみられる[Van de Laarら2007]。
- 脳のMRI:斜台低形成[de Geusら2018],小脳虫部低形成[Donovanら2017]
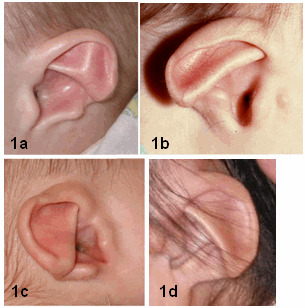
図1:耳
1a:摘み取られたような耳輪。
突出した対耳輪が外側の耳輪にまで達するが、対耳輪は対珠(訳注:本文では「耳珠」となっているが、本文のほうが誤りで、この図の説明のほうが正しいものと思われる)とはつながらない。
耳垂は欠損している。
1b:対耳輪は対珠とはつながっておらず、耳垂はきわめて小さい。
耳前部副耳が時に現れる。
1c:摘み取られたような耳輪と突出した対耳輪。
対耳輪は耳輪縁にまで達するが、対珠とはつながらない。
三角形の耳甲介。
耳垂は欠損している。
1d:薄く、折れ込みのない耳輪と突出した対耳輪下脚。
対耳輪下脚と対珠の間には切れ込みがある。
痕跡状の耳垂。
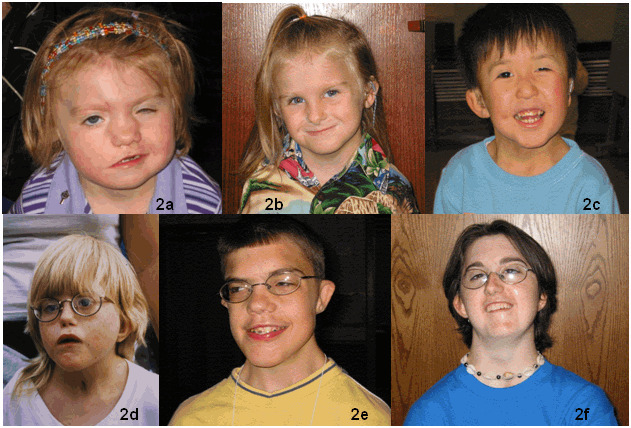
図2:顔
2a:2歳半の女児。
四角い顔、丸い眼、鼻根の広い平坦な鼻、片側の顔面神経麻痺。
2b:軽度型のCHARGE顔貌を呈する5歳の女児。
やや四角い顔、目立つ鼻柱。
なで肩に注目。
2c:7歳の男児。
四角い顔、広めの鼻根。
折れ込みのない耳輪を伴う突出した耳と広い頸に注目。
2d:9歳の女児。
四角い顔、丸い眼、広い頸、なで肩。
両側の顔面神経麻痺のため、表情がないことに注目。
2e:15歳の男児。
長いながらもやや四角い顔、広い頸、なで肩に注目。
2f:18歳の女性。
四角く非対称な顔、突出した耳、後傾した頭、広い頸、なで肩。

図3:手
典型的なCHARGEの手。
四角い手、短い第2-第5指、第2-5指の形に似た拇指、ホッケースティック形の屈曲線。
家族歴
常染色体顕性遺伝に一致した家族歴がみられる。
CHD7疾患罹患者の大半は孤発例(新生のCHD7の病的バリアントの結果、家系内で1人だけ発生した状態)であるものの、常染色体顕性遺伝に一致して現れた家族例や、生殖細胞系列モザイクの報告がみられる[Bergmanら2011b,Legendreら2017]。
注:CHD7疾患に合致する症候の家族歴がないからといって、CHD7疾患ではないということにはならない。
診断の確定
発端者におけるCHD7疾患の診断は、CHD7疾患を示唆する臨床所見、画像所見を有することに加え、分子遺伝学的検査によりCHD7のヘテロ接合性病的バリアントが同定されることにより確定する(表1参照)。
注:CHD7に意義不明のバリアントのヘテロ接合が検出された場合、それはCHD7疾患の診断を確定するものでも否定するものでもない。
分子遺伝学的検査としては、表現型に応じて、遺伝子標的型検査(CHD7の単一遺伝子検査、マルチ遺伝子パネル)と網羅的ゲノム検査(染色体マイクロアレイ、エクソームシーケンシング、エクソームアレイ、ゲノムシーケンシング)を組み合わせて用いることが考えられる。
遺伝子標的型検査の場合は、まず臨床医のほうで、関与が疑われる遺伝子の目星をつけておく必要があるが、網羅的ゲノム検査の場合、その必要はない。
CHD7疾患を示唆する症候を有する例については、遺伝子標的型検査を用いて診断を行うことになるものと思われる(「方法1」参照)。
一方、非定型的な症候を示す例については、ゲノム検査で診断が行われることになるものと思われる(「方法2」参照)。
方法1
単一遺伝子検査
CHD7の配列解析は、遺伝子内の小さな欠失/重複、ならびにミスセンス・ナンセンス・スプライス部位バリアントの検出を目的として行われる。
注:用いる配列解析の手法によっては、1エクソン、複数エクソン、遺伝子全体といったレベルの欠失/重複は検出されないことがある。
最初に用いた配列解析でバリアントが検出されなかった場合、次に実施すべき手法は、エクソン単位あるいは遺伝子全体の欠失/重複を検出するための遺伝子標的型欠失/重複解析、ないし、遺伝子全体の欠失を検出するための染色体マイクロアレイ(CMA)である。
マルチ遺伝子パネル
意義不明のバリアントの検出、ならびに、いま問題にしている表現型と無関係な遺伝子の病的バリアントの検出を抑えつつ、疾患の原因となった遺伝子を最も安価に同定しうるのは、CHD7その他の関連遺伝子(「鑑別診断」の項を参照)を含むマルチ遺伝子パネル(例えば、発達遅延用、コロボーマ用、難聴用、心奇形用、Kallmann症候群用、正常嗅覚型低ゴナドトロピン性性腺機能低下用のもの)であろうと思われる。
注:
(1)パネルに含められる遺伝子の種類、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、今このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。
(3)検査機関によって、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝子検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。方法2
網羅的ゲノム検査の場合、あらかじめ臨床医のほうで関与が疑われる遺伝子の目星をつけておく必要はない。
エクソームシーケンシングが最も広く行われているものの、ゲノムシーケンシングを行うことも可能である。
CHD7疾患では、一般に複数の奇形が生じるため、CHD7疾患の典型的症候(CHARGE症候群の表現型)が一見して明らかということでなければ、染色体マイクロアレイ検査を最初に行うことも合理的である。
あるいは、エクソームシーケンシングで診断がつかないような場合は、配列解析では検出できない単一エクソンないし複数エクソンの欠失/重複を検出するためのエクソームアレイ(もしこれを行うことが可能であれば)も検討に値するかもしれない。
網羅的ゲノム検査の基礎的情報についてはここをクリック。
ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
表1:CHD7疾患で用いられる分子遺伝学的検査
| 遺伝子1 | 手法 | その手法で病的バリアント2が 検出される発端者の割合3 |
|---|---|---|
| CHD7 | 配列解析4 | 98% |
| 遺伝子標的型欠失/重複解析5,6 | 2% |
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- この遺伝子において検出されているバリアントの情報については、「分子遺伝学」の項を参照。
- 割合を示す数字は、locus-specific database CHD7.org[Janssenら2012]より引用。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。
バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。 - 遺伝子標的型の欠失/重複解析では、遺伝子内の欠失や重複が検出される。
具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失ないし重複の検出を目的に設計された遺伝子標的型マイクロアレイなどがある。 - 現在までに検出されている欠失の大部分は遺伝子全体の欠失で、これは遺伝子標的型の欠失/重複解析で検出可能である。
ただ、この手法では、CHD7や隣接遺伝子まで含んだより大きな欠失にまでは対応することはできない。
染色体マイクロアレイ解析を用いれば、そうした欠失についても検出が可能であろう。
臨床的特徴
臨床像
分子レベルになる前の時代においては、コロボーマ、心疾患、後鼻孔閉鎖、成長発達遅滞、生殖器の低形成、耳の奇形(難聴を含む)といった原因不明の臨床症候の組合せを表現するためのCHARGEという頭字語の使用が提唱された[Pagonら1981]。
その後、この疾患はCHARGE連合と呼ばれるようになり、それに対する改良型臨床診断基準が確立した[Blakeら1998,Verloes 2005]。
CHD7のバリアントや欠失のヘテロ接合がCHARGE症候群を引き起こすということが発見された[Visserら2004]後、CHARGE症候群の発端者の家族に対して分子遺伝学的検査が行われるようになったことで、表現型のスペクトラムは、以前にCHARGE症候群の臨床診断基準として提唱されたものを満たさない表現型を含むところにまで拡大することが明らかとなった[Lalaniら2006,Delahayeら2007,Jongmansら2009,Bergmanら2011b,Haleら2016]。
このことからわかるように、CHD7疾患は、同一家系内の罹患者間、ならびに同一の病的バリアントを共有する別家系の罹患者間にあってさえ、臨床症候の現れ方に大きなばらつきがみられる疾患である[Jongmansら2008]。
ここからの節では、罹患者がCHD7の病的バリアントを有していることが確認できたとする報告のみを取り扱う。
今日に至るまで、CHD7疾患で症候が単発であったとする報告は非常に少ないことに加え、その多くが、CHD7疾患の表現型スペクトラムに含まれる他の症候に関する十分な精査を行った上での報告になっていない。
こうしたことから、表2に示した割合の数字(分子レベルの確認がなされたCHARGE症候群の罹患者から得られた数字[van Ravenswaaij-Arts & Martin 2017])は、マルチ遺伝子パネル検査やゲノム検査でCHD7の病的バリアントが確認された罹患者の詳しい臨床的評価が積み上がっていくに従って、変化していく可能性が高い(表5参照)。
表2:CHARGE症候群と確認された罹患者に現れるCHD7疾患の症候
| 症候 | その症候を有する罹患者の割合 | コメント | |
|---|---|---|---|
| 眼のコロボーマ(網膜の小さなコロボーマから無眼球まで幅あり) | 80% | 光過敏症、屈折異常、上方視野・中心視野の欠損、失明 網膜剥離のリスク上昇 |
|
| 後鼻孔閉鎖/狭窄 | 45% |
|
|
| 脳神経の機能障害/奇形 | Ⅰ:嗅覚減退/脱失 | 90% | 嗅覚減退/脱失は、低ゴナドトロピン性性腺機能低下の予兆となる。 |
| Ⅶ:顔面神経麻痺 | 40% |
|
|
| Ⅷ:感音性難聴/前庭機能障害 | 95%超 |
|
|
| Ⅸ/Ⅹ:吸啜・嚥下、消化管運動性異常 | 60%-80% |
|
|
| 耳の奇形 | 耳介奇形 | 90% |
|
| 耳小骨奇形 | 80% |
|
|
| Mondini型奇形 | 90% | 特に高音域の感音性難聴 | |
| 半規管奇形 | 94% | 平衡覚や視覚処理に影響し、運動発達遅延に発展。 | |
| 口唇裂口蓋裂 | 25%-50% | ||
| 内分泌1 | 低ゴナドトロピン性性腺機能低下 | 50%-70% |
|
| 成長障害 | 70% | 成長ホルモン分泌不全に起因するものあり(10%以下) | |
| 甲状腺機能低下 | 15%-20% | ||
| 発達遅滞/知的障害 | 90%超/60% | 感覚(聴覚,視覚,平衡覚)障害、病気、入院に起因する発達遅延 | |
| 心血管系奇形 | 74% |
|
|
| 気管食道奇形 | 20% | 気管食道瘻,喉頭気管軟化症,胃食道逆流症を伴う、あるいは伴わない食道閉鎖が原因となって、哺乳障害,呼吸困難,誤嚥(性肺炎),副鼻腔炎を発症。 | |
| 脳 | 斜台低形成 トルコ鞍低形成/J字形トルコ鞍 |
95% | |
| その他 | 50% | 小頭症,脳室拡大,Dandy-Walker奇形,脳梁低形成(30%),小脳虫部低形成(50%),脳幹ないし前頭葉の低形成 | |
| 癲癇 | 30% | 発症は全年齢に分布し、最も多いのは全身性強直性間代性痙攣と欠神癲癇 | |
| 腎奇形 | 30% |
|
|
CHARGE症候群の典型例と部分発現例の両方を含むものの、すべて分子レベルで確認済の罹患者を用いたもの[van Ravenswaaij-Arts & Martin 2017]。
注:本表に示した数字は、確認過程(すなわち、分子遺伝学的検査に至った理由)によって大きく変わってくるものである。
マルチ遺伝子パネル検査やゲノム検査がますます一般的になってきていることから、今後は、典型的なCHARGE症候群の症候を有しない多くの非定型罹患者がCHD7疾患と診断されていくようになるものと思われる
- Balasubramanian & Crowley[2017],Xuら[2018]
CHD7の病的バリアントを有する罹患者の大多数は、典型的なCHARGE症候群の症候、あるいはCHARGE症候群に似た症候を示すことから、以下に述べる臨床症候は、CHD7疾患罹患者の大半に当てはまるものである。
一方、CHD7の病的ミスセンスバリアントに起因して、嗅覚脱失を伴う、あるいは伴わない単発性の低ゴナドトロピン性性腺機能低下が生じる例は稀であるように思われる[Xuら2018]。
発達
前庭奇形に起因して、運動機能の遅延は常にみられ、頸のすわりの悪さ、5点這い(訳注:おそらく、両手足だけでなく、胸や腹も床についたハイハイを指すものと思われる)、運動発達指標の遅れ、微細運動技能の遅れといった形で現れる。
難聴、視力低下、前庭奇形、病気や入院、認知障害に起因して、言語発達に遅れが生じる。
複数の感覚障害(視覚,聴覚,平衡覚,嗅覚)を伴うため、認知機能の評価は困難で、運動やスピーチ/言語の遅れの多くは、こうした感覚障害に伴う二次性のものである。
それでもなお、CHARGE症候群の臨床症候を有する罹患者の50%は、知能が正常範囲内にある[Vesseurら2016b]。
運動能力が低く、多くの医学的問題を抱える子どもに比べ、歩行能力が高く、医学的問題の少ない子どものほうが優れた適応行動能力を示す[Salem-Hartshorne & Jacob 2005]。
行動学的症候としては、注意欠陥多動性障害、反復行動、強迫性障害がしばしば報告されている。
自虐行動もしばしばみられる。
疼痛閾値が高いため、子どもは、往々にして他者から攻撃的と誤解される行動をとりがちである[Hartshorneら2005]。
CHARGE症候群の臨床症候を有する成人の多くは自立した生活を送り、学士あるいはそれ以上の学位をもつ人も多い。
しかし、個々の罹患者ごとに、有している症候、個々のニーズに対応して用意された教育プログラム、利用可能な教育資源といったものに違いがみられるため、自立のレベルには大きな幅がある[Blakeら2005,Hartshorneら2016]。
他の症候
消化器系
胃食道逆流症、便秘、腹痛といった、主として消化管運動性に関連する消化器系の問題がしばしばみられる。
哺乳困難により、しばしば経管栄養となったり、誤嚥の問題が生じたりする。
遅発性の問題としては、腸回転異常、腸重積、食物の口への詰め込み過ぎによる窒息などがある[Hudsonら2015,Blake & Hudson 2017]。
免疫不全
胸腺の欠損(稀)ないしT細胞の数や機能の低下に起因する免疫不全が生じることがある[Wongら2015b]。
反復性の上気道感染症が多くみられる。
骨格系
骨格系の問題としては、頭蓋骨早期癒合症、椎骨奇形、脊柱側彎(これは罹患者の大多数でみられる)[Doyle & Blake 2005]、肋骨の過不足、長骨の欠損(稀)、欠指趾、多指趾、第2-5指の形に似た拇指、短指趾(これは多くみられる)などがある[Van de Laarら2007]。
関節の過可動性や拘縮も、本症候群の症候の一部である。
神経筋系
CHARGE症候群では、神経筋の問題が高頻度にみられ、そのうち最も多いのが筋緊張低下(これにより脊柱側彎が生じる)と肩甲帯の筋の異常である[O‘Gradyら2016]。
固有受容が減退し、平衡覚の問題と相まって、しばしば無理な姿勢(pressure-building posture)(上下逆の姿勢や両脚を絡ませる姿勢)を好んでとるようになる[Brownら2005]。
歯
歯の問題としては、オーバーバイトの過大、部分性無歯症、石灰化不全などがある[Chettyら2020]。
寿命
CHD7疾患にみられる表現型は非常に広い幅をもつため、寿命は症候の重症度によって大きく左右されることになる。
出生時に重度の異常(特に複合心奇形)があり、気道や摂食哺乳の問題が絡んでくるような場合、最初の数年間の死亡率は高くなる。
摂食哺乳障害は、多くの場合、脳神経の異常に起因して生じるが、その後、次第に改善していく。
複雑な手術を何度も行わざるをえないことと、CHARGE症候群で報告されている呼吸の問題や麻酔の難しさ[Blakeら2009]が相まって、医療行為に付随するリスクが高まることになる。
最初の2、3年を過ぎた後も、死亡率(罹病率や身体的脆弱性も含め)は高い状態のままで推移し、親のほうから頻回の病気、感染、入院の報告がある[Bergmanら2010]。
小児期、思春期、成人期の死亡率の高さについては、おそらく、心奇形の影響、感染症、誤嚥や窒息[Corsten-Janssenら2016]、閉塞性・中枢性の無呼吸を含む呼吸の問題に加え、おそらく癲癇発作も関与しているものと思われる。
多くの家系で、腸捻転[Lai & Feng 2006]や腸重積といった重大な(時として致死的な)腸の問題が報告されている。
こうした合併症があるとはいえ、罹患者の多くについては、正常な寿命を有する。
CHARGE症候群の臨床症候を有する60歳代の罹患者で、健康状態が良好な例もみられる。
遺伝型と表現型の相関
CHD7関連CHARGE症候群については、遺伝型と表現型との間に明確な相関関係はみられない[Legendreら2017]。
ただ、原則的にとまでは言えないものの、一般に、ミスセンスバリアントの例では表現型の重症度が低めのようである[Bergmanら2012]。
CHD7関連の、嗅覚脱失を伴う、もしくは伴わない低ゴナドトロピン性性腺機能低下については、ナンセンスバリアントではなくミスセンスバリアントが関わっている可能性が高い。
発生頻度
ゲノム検査が広く行われるようになってきている現況から、CHD7疾患の発生頻度を推定することは現時点では困難である。
以前、CHARGE症候群の診断が臨床症候や遺伝子特異的な分子検査に基づいてなされていた頃の推定発生頻度について言うと、新生児15,000人に1人とするオランダの数字[Janssenら2012]と、8,500人に1人とするカナダの数字[Issekutzら2005]の間にあるものと推定されていた。
遺伝的に関連のある疾患(同一アレル疾患)
CHD7の生殖細胞系列の病的バリアントに関しては、このGeneReviewで述べているもの以外の表現型の存在は知られていない。
鑑別診断
CHD7疾患と一部の症候が重なる遺伝性疾患を、表3、表4にまとめて示した。
表3:CHD7疾患との鑑別診断において検討を要する遺伝子
| 遺伝子 | 疾患名 | 遺伝形式 | 鑑別すべき疾患にみられる臨床症候 | |
|---|---|---|---|---|
| CHD7疾患と重なる症候 | CHD7疾患と異なる症候 | |||
| 34以上の遺伝子 | Joubert症候群 | 常染色体潜性 X連鎖性 2遺伝子性 |
両側性の脈絡膜網膜コロボーマ,腎不全に移行する腎間質性線維症,肝線維症,新生児多呼吸,小脳虫部無形成/低形成,多指趾 | 神経画像におけるmolar tooth sign,特徴的X線像,CHD7疾患にみられる形態異常の欠如 |
| EYA1 SIX1 SIX5 |
鰓耳腎スペクトラム障害 | 常染色体顕性 | 聾,外耳奇形,外側半規管低形成,腎奇形 | 鰓性瘻と鰓性嚢胞,CHD7疾患にみられる形態異常の欠如 |
| KDM6A KMT2D |
Kabuki症候群 | 常染色体顕性 X連鎖性 |
口蓋裂,心奇形,時にみられるコロボーマ,難聴,成長障害 | 特徴的顔貌:下眼瞼外側1/3の反転を伴う長い眼瞼裂,疎な眉,突出した大きな耳(これらは年齢とともに顕著化),指腹隆起 |
| PAX2 | PAX2疾患 (腎コロボーマ症候群) |
常染色体顕性 | 網膜/視神経コロボーマ,腎奇形,時にみられる難聴 | 多発性先天奇形の欠如 |
| BMP4 | 症候群性小眼球症6型 (OMIM 607932) |
常染色体顕性 | コロボーマ,外耳奇形,難聴,心奇形,生殖器低形成,口唇裂口蓋裂,下垂体異常,腎奇形 | CHD7疾患より無眼球の頻度が高い。 |
| EFTUD2 | 小頭症を伴う下顎顔面異骨症 | 常染色体顕性 | 後鼻孔閉鎖,外耳奇形,難聴,先天性心疾患,成長障害,口唇裂口蓋裂,食道閉鎖 | 頰骨低形成に起因する特徴的な頭蓋顔面形態 |
| FGFR1 | FGFR1 Kallmann症候群1(OMIM 147950) | 常染色体顕性 | コロボーマ,難聴,生殖器低形成,嗅覚減退/脱失,口唇裂口蓋裂1 | |
| GLI2 | Culler-Jones症候群 (OMIM 615849) |
常染色体顕性 | 外耳奇形,難聴,口唇裂口蓋裂,成長障害,下垂体異常,腎奇形 | 中顔面低形成と軸後性多指趾 |
| GLI3 | Pallister-Hall症候群 | 常染色体顕性 | コロボーマ,外耳奇形,先天性心疾患,成長障害,生殖器低形成,口唇裂口蓋裂,下垂体異常,腎奇形 | 視床下部過誤腫,中央列多指趾 |
| JAG1 NOTCH2 |
Alagille症候群 | 常染色体顕性 | 先天性心疾患,腎奇形 | 胆汁鬱滞,蝶形椎,後部胎生環,逆三角の顔 |
| MYCN | Feingold症候群1型 | 常染色体顕性 | 難聴,心奇形,食道閉鎖,腎奇形 | 中節骨短縮 |
| OTX2 | 症候群性小眼球症5型 (OMIM 610125) |
常染色体顕性 | コロボーマ,成長障害,生殖器低形成,口蓋裂,下垂体異常 | CHD7疾患より無眼球の頻度が高い。 脳梁無形成。 耳頭-異下顎複合に似る。 |
| POLR1C POLR1D TCOF1 |
Treacher Collins症候群 | 常染色体顕性 常染色体潜性 |
後鼻孔閉鎖,外耳奇形,難聴,口蓋裂 | 頰骨低形成に起因する特徴的頭蓋顔面形態,正常な知能 |
| SOX2 | SOX2疾患 | 常染色体顕性 | コロボーマ,難聴,先天性心疾患,成長障害,生殖器低形成,食道閉鎖,下垂体異常 | CHD7疾患より無眼球の頻度が高い。 |
| TBX22 | Abruzzo-Erickson症候群 (OMIM 302905) |
X連鎖性 | コロボーマ,難聴,成長障害,口蓋裂 | 橈尺骨癒合,突出した大きな耳 |
| ZEB2 | Mowat-Wilson症候群 | 常染色体顕性 | 外耳奇形,先天性心疾患,成長障害,生殖器低形成,口蓋裂,腎奇形 | 特徴的顔貌(持ち上がった耳垂を含む),Hirschsprung 病,男性の尿道下裂 |
- 嗅覚の鈍麻や脱失がみられる場合には、Kallmann症候群、特にFGFR1の病的バリアントに起因して生じるKallmann症候群を疑う必要がある(Kallmann症候群には、CHD7を含む16の遺伝子の病的バリアントが関与していることが知られている)。
表4に、CHD7疾患と症候が顕著に重なる染色体異常症候群を掲載した。
CHD7疾患と最低2,3の症候が重なる染色体欠失症候群は、他にも多く存在する。
表4:CHD7疾患と症候が顕著に重なる染色体異常症候群
| 遺伝的機構 | 疾患名 | 鑑別すべき疾患にみられる臨床症候 | |
|---|---|---|---|
| CHD7疾患と重なる症候 | CHD7疾患と異なる症候 | ||
| 22q11.2の欠失 | 22q11.2欠失症候群 | 先天性心疾患,口蓋異常1,学習障害,免疫不全,重大な摂食哺乳障害,腎奇形,難聴,喉頭気管食道奇形,成長ホルモン分泌不全,骨格の異常 | 半規管の異常はほとんどみられない。眼のコロボーマも稀。CHD7疾患より早期に摂食哺乳障害が解消,形態異常(顔,耳,手)の様相が異なる |
| 22q11の逆位重複 | キャットアイ症候群(OMIM 115470) | 虹彩コロボーマ,肛門瘻を伴う鎖肛,耳前部の副耳ないし小窩,心奇形・腎奇形の頻発 | CHD7疾患でみられる形態異常や耳科的症候の欠如 |
(訳注:表中に振られている「1」の注が欠落している模様。)
VACTERL連合(OMIM 192350)
CHD7疾患とVACTERL連合は、ともに椎骨奇形、心奇形、気管食道瘻(ないし食道閉鎖)、腎奇形、四肢奇形を症候として共有する。
ただ、鎖肛は、VACTERL連合では多くみられるが、CHD7疾患では稀である。
逆に、典型的なCHD7疾患でみられる側頭骨の奇形、後鼻孔閉鎖、特徴的な耳の症候(外耳ならびに内耳)、脳神経奇形は、VACTERL連合ではほとんど報告がない。
VACTERL連合の遺伝的基礎については、まだよくわかっていない。
出生前の催奇形因子への曝露に伴って二次的に生じるCHARGE症候群類似の症候
妊娠の第1三半期のいずれかの時期にAccutaneTMに曝露すると、神経堤細胞の遊走の異常に伴う奇形が生じることがある。
具体的には、小耳症/無耳症、小下顎症、口蓋裂、心臓の円錐動脈幹奇形、大動脈弓奇形、胸腺の欠損、網膜や視神経の異常、中枢神経系奇形などがある[Lammerら1985]。
抗甲状腺薬、中でも特にメチマゾールに曝露すると、後鼻孔閉鎖、食道閉鎖、虹彩や網膜のコロボーマ、難聴、神経発達遅延をはじめとする種々の先天奇形が生じることが報告されている。
妊娠の第1三半期にこれに曝露した場合の胎児の先天異常発生リスクは、2-3%と推定されている[Andersenら2013,Komoikeら2013,Andersenら2017]。
臨床的マネジメント
CHD7疾患の症候に対する管理は複雑になることがあり、臨床医、各分野の専門療法士、教育者などを含む多専門の共同管理体制が必要になる可能性がある。
CHARGE症候群の臨床的管理のためのガイドライン(1ページの要約付き)[Triderら2017](図4参照)と、頭蓋の画像作成のガイドライン[de Geusら2017](訳注:本文中にはないが、「図5」のことを指すものと思われる。)(全文)が公開されている。
図4:CHARGE症候群のチェックリスト:生涯の健康管理(頭から足先まで)
(訳注:実質的には「表」であるが、原文では全体が写真になっているため、「図」となっている。)
※色付きの枡は、評価を行うべき重要な時期であることを示す。
| 乳幼児期 (0-2歳) |
小児期 (3-11歳) |
思春期 (12-17歳) |
成人期 (18歳-) |
||
| 遺伝 | 臨床診断(Blakeら、もしくはVerloesらの基準) | ||||
| 遺伝検査―遺伝科紹介(CHD7の解析,アレイCGH) | |||||
| 遺伝相談 | |||||
| 神経 | 中枢神経系奇形/嗅球低形成/側頭骨[半規管]奇形―MRI/CTが必要 | ||||
| 癲癇発作―年齢が高いほうが多い―脳波を検討 | |||||
| 脳神経の問題―嗅覚脱失,顔面神経麻痺,感音性難聴,めまい,嚥下障害のモニター | |||||
| 眼 耳 鼻 喉 |
コロボーマ,網膜剥離のリスク―眼科紹介(乳幼児期に散瞳検査,視力検査) | ||||
| 角膜露出―潤滑用点眼薬 | |||||
| 羞明―着色眼鏡,日よけ帽 | |||||
| 後鼻孔閉鎖/口蓋裂/気管食道瘻―耳鼻科,形成外科紹介 | |||||
| 聴力検査とティンパノメトリー,反復性耳感染症のモニター | |||||
| 盲聾者に対する適応サービス | |||||
| できるようであれば人工内耳の評価 | |||||
| 閉塞性睡眠時無呼吸―扁桃/アデノイド肥大のモニター | |||||
| 分泌過多―Botox,投薬を検討 | |||||
| 歯の問題―麻酔下でのクリーニングを検討 | |||||
| 心臓 呼吸 |
心奇形が多い―大/小奇形,血管輪や不整脈の可能性(心エコー,胸部X線,心電図)―循環器科紹介 | ||||
| 上顎洞炎,肺炎,喘息―モニター | |||||
| 麻酔のリスク(挿管困難/術後の気道閉塞/誤嚥)―詳細な術前評価,手術をまとめて行う | |||||
| 消化 泌尿生殖 |
胃食道逆流症―消化器科紹介―プロトンポンプ阻害薬を用いた運動性改善薬物療法を検討 | ||||
| 吸啜/咀嚼/嚥下障害―摂食チームの評価/介入 | |||||
| 誤嚥リスク,気管食道瘻―嚥下検査 | |||||
| 補助食必要か―しばしば胃瘻チューブや胃空腸瘻チューブ必要 | |||||
| 便秘―センナグリコシドとポリエチレングリコールを検討 | |||||
| 腎奇形―腹部超音波+/-排尿時膀胱尿道造影,血圧モニター | |||||
| 内分泌 | 低ゴナドトロピン性性腺機能低下―黄体形成ホルモン,卵胞刺激ホルモンを3ヵ月間 | ||||
| 生殖器低形成(停留精巣あり―精巣固定術を検討) | |||||
| 思春期遅発―内分泌科紹介―ゴナドトロピン値,ホルモン補充療法 | |||||
| 骨粗鬆症―骨密度測定 | |||||
| 成長不良―内分泌科紹介―成長ホルモン刺激試験,成長ホルモン治療 | |||||
| 肥満―モニター | |||||
| 受胎能力と避妊―面談 | |||||
| 免疫 | 開心術の際に胸腺の存在を注視 | ||||
| 通常の予防接種/思春期には抗体価測定 | |||||
| 反復性感染症―免疫科紹介 | |||||
| 筋 骨 |
脊柱側彎/後彎―モニター | ||||
| 運動能力(運動失調,筋緊張低下の影響)―評価 | |||||
| 心理 発達 |
粗大/微細運動技能の評価―作業療法,理学療法 | ||||
| コミュニケーション,言語,筆記能力―言語療法 | |||||
| 盲聾コンサルタントを検討 | |||||
| 学校,状況,場所,体制への移行準備 | |||||
| 心理教育的評価,個別的教育計画 | |||||
| 睡眠障害―メラトニンを検討 | |||||
| 行動管理―自己制御,衝動コントロール,不安,執着,強迫,怒り | |||||
| トイレ使用能力―支援 | |||||
| 生活能力/適応行動/社会的技能/社会的遊び | |||||
| 性的なことへの準備 | |||||
| 家族のストレス―支援ならびに情報提供 | |||||
| 医療の自己管理―服薬管理,身体状況把握,医療機関への単独での受診 | |||||
初期診断後の評価
CHD7疾患と診断された罹患者については、疾患の及んでいる範囲や治療のニーズを把握するため、診断に至るまでの評価の一部として、もしまだ行っていないようであれば、表5に要約したような評価を行うことが推奨される。
表5:最初の診断に続いてCHD7疾患の罹患者に対して行うことが推奨される評価項目
| 系/懸念事項 | 評価項目 | コメント |
|---|---|---|
| 体格 | 身長,体重,頭囲の計測 | 成長障害や肥満のチェック目的 |
| 眼 | 眼科的評価 |
|
| 聴覚 平衡覚 |
聴覚評価1 | 伝音性ならびに感音性難聴のチェック |
| 平衡覚に関する臨床評価 | ||
| 側頭骨のMRI/CT | 半規管,蝸牛,脳神経の評価(詳しくは図5を参照) | |
| 鼻 喉 |
後鼻孔閉鎖/狭窄の臨床評価 |
|
| 気管食道瘻のチェック | 下記の場合のみ行う
|
|
| 扁桃とアデノイド | 閉塞性無呼吸がみられた場合に行う。 | |
| 口 | 口蓋裂,粘膜下口蓋裂,鼻咽腔閉鎖機能不全の臨床評価 | 頭蓋顔面チームへの紹介を検討 |
| 歯科医による開始時点の評価を、ふつう3歳まで(口蓋裂があるときはそれ以前)に行う。 |
|
|
| 心血管 | 心電図と心エコー3 | 必要性があれば心臓病専門医へ紹介 |
| 呼吸器 | 睡眠ポリグラフの検討 | 睡眠時無呼吸のチェックのため |
| 肺機能検査の検討 | 重度の脊柱側彎があり、拘束性肺疾患の可能性のある年長者に対して行う。 | |
| 消化器 摂食 |
嚥下障害や誤嚥の徴候,症状のチェック | 誤嚥や肺炎の疑われる例については、ビデオ嚥下造影検査,ならびに栄養摂食チームによる評価を検討 |
| 胃食道逆流症や消化管運動性のチェック4 | 必要性があれば消化器専門医へ紹介 | |
| 泌尿器 生殖器 |
男性:小陰茎や停留精巣のチェック |
|
| 女性:骨盤の超音波検査を検討 |
|
|
| 筋骨格 | 脊柱側彎の臨床的チェック | ・開始時点の記録としての脊椎のX線写真を検討 ・整形外科医への紹介を検討 |
| 神経 | 脳神経奇形の臨床的チェック |
|
| 癲癇発作が疑われるときは頭蓋のMRIと脳波 | 神経内科医への紹介を検討 | |
| 発達 | 発達のチェック |
|
| 精神 行動 |
神経精神医学的評価を検討 |
|
| 内分泌 |
|
嗅覚脱失がゴナドトロピン放出ホルモンの機能異常のサインである可能性あり。 |
|
||
| 腎 | 腎超音波検査 | 腎奇形,水腎症,腎石灰化のチェック目的 |
| 血圧測定 | 高血圧のチェック目的 | |
| 免疫 | 免疫学的評価8や免疫内科医への紹介を検討 | 反復性ないし原因不明の感染を伴う例に行う。 |
| 遺伝カウンセリング | 遺伝の専門家9が行う | 医学的、個人的決断が行いやすくなるよう、本人や家族に対し、CHD7疾患の本質、遺伝形式、意味について情報提供を行う。 |
| 家族への支援/情報資源 |
|
- 聴覚評価についての詳細については、「遺伝性難聴・聴力喪失概説」のGeneReviewを参照。新生児聴覚スクリーニングで正常と出たときに、聴覚評価を併せて行う。
- Chettyら[2020]
- 不整脈、ならびに血管奇形を含む心臓の構造異常を評価するために行う[Corsten-Janssenら2013]。
- その他、評価すべきものとして、腸回転異常、便秘、慢性の腹痛、腹部膨満感、後期ダンピング症候群などがある[Morganら2017]。
- de Geusら[2017]
- 具体的には、黄体形成ホルモン、卵胞刺激ホルモン、性ホルモン(男性では総テストステロン、女性ではエストロゲン)の血清濃度測定。
- 思春期ならびに成人期に行う。
- 具体的には、反復性感染の既往がある場合、免疫グロブリン値(IgG,IgM,IgA)ならびにT細胞サブセット、B細胞サブセットなどがある
- 臨床遺伝医,認定遺伝カウンセラー,認定上級遺伝看護師をいう。
「単発性ゴナドトロピン放出ホルモン(GnRH)分泌不全症」のGeneReviewも参照。
症状に対する治療
CHD7疾患の子どもの管理にあたっては、各専門科間の協調のとれたケアが必要となる(表6)。
| 症候/問題 | 治療 | コメント/その他 |
|---|---|---|
| 成長不全 | 本表中の「嚥下障害/栄養不良」を参照 |
|
| 肥満 | 体重負荷運動ならびに食事介入 | 学校体育における適応;これは感覚にも働く |
| 視力低下/盲 | 屈折矯正用レンズ/標準治療 | 早期介入もしくは学区を通じた地域の視覚障害者サービス;利用可能なら聾-盲者サービス |
| 角膜露出 | 潤滑用点眼薬 | |
| 羞明 | 着色レンズもしくは日よけ帽 | |
| 難聴/聾 | 難聴の程度に応じた感音性難聴と伝音性難聴の治療 |
|
| 人工内耳 | 人工内耳手術の術前評価の一部として、CTによる骨の基準点の把握と、MRIによる前庭・顔面神経の把握 | |
| 骨導補聴器 | 中耳の骨の異常により中耳に問題があるものの、蝸牛神経は健全である場合に用いる。 | |
| FM補聴器 | 学齢期の子どもに用いる。 | |
| 慢性中耳炎 | 鼓膜チューブ挿入 | ティーン世代まで何度か入れ替える必要あり。 |
| 平衡覚障害 | 体幹を支えるため教室や診療室の調整(仰臥位を含む) | 頻回の休息を要することがある。 必要に応じ、作業療法、理学療法、姿勢定位・運動性のサービスに紹介 |
| 平衡覚を鍛えるための空手その他のプログラム | ||
| 筋膜リリースの検討 | 姿勢や柔軟性が改善される可能性あり | |
| コミュニケーション | 聴覚と視覚の低下の程度によって変わる。 | 出生後、可能な限り早期に地域の聾-盲サービスや州の聾-盲プロジェクトに相談。 |
| 後鼻孔閉鎖/狭窄 | 乳児:気管切開や気管内挿管による気道バイパス作製 | 気道の開存性を維持するには複数の手術/拡大が必要。 |
| 外科的修復 | ||
| 食道閉鎖/気管食道瘻 | 外科的修復の標準治療 | |
| 口蓋裂 | 外科的修復 | 頭蓋顔面チームの手で行うのが理想的。 |
| 齲歯/部分性無歯症 | 歯科治療に鎮静が必要なことあり。 | 「避けるべき薬剤/環境」の項を参照。 |
| 分泌過多 | グリコピロレートやBotox®などの標準処方 | |
| 先天性心疾患/不整脈/起立性頻脈症候群 | 心臓病専門医による治療 | |
| 閉塞性睡眠時無呼吸 | 扁桃摘出術、アデノイド切除術を検討 | 時にCPAPが有用。 |
| 睡眠障害 | 睡眠衛生;メラトニンの試用を検討 | 睡眠障害専門医への紹介を検討 |
| 嚥下障害/栄養不良 | 経鼻胃管ないし胃瘻チューブが必要なことあり | 嚥下障害の臨床的兆候や症候がみられるときは、摂食に関する臨床評価やX線による嚥下検査を躊躇なく行う。 |
| 胃食道逆流症:標準治療 | ||
| 便秘 | 刺激性下剤や浸透圧性緩下剤などの処方による治療 |
|
| 停留精巣 | 泌尿器科医による標準治療 | |
| 重度の脊柱側彎 | 整形外科医による標準治療 | 骨格ないし神経筋のほうに原因があることあり。 |
| 発作性障害 | 神経内科医による標準治療 | 多くの場合、標準的な抗けいれん薬が有効 |
| 発達遅滞/知的障害 | 「発達遅滞/知的障害の管理に関する事項」の項を参照 | 可能であれば聾-盲サービスを利用する。 |
| 行動の問題 | 強迫性障害,広汎性発達障害:まずマインドフルネス認知療法(心理士と聾-盲の専門家)を行い、その後、投薬を考える。 |
|
| 注意欠陥多動性障害:適切なコミュニケーション法を確立するとともに、安全な環境下で診察のための刺激を十分に与える。 | 投薬よりこのほうが有用なことあり。 | |
| 低ゴナドトロピン性性腺機能低下 | 内分泌内科医による標準治療 | ふつう、骨粗鬆症を含む全身の健康のことを考慮した思春期誘導のためのホルモン補充療法が行われる。 「単発性ゴナドトロピン放出ホルモン分泌不全症」のGeneReviewを参照。 |
| 甲状腺機能低下 | 内分泌内科医による標準治療 | |
| 腎奇形 | 泌尿器科医による標準治療 | |
| 高血圧 | 標準治療 | |
| 反復性感染症 | 免疫専門医による標準治療 | ワクチンの再接種が必要なことあり。 |
| 家族のニーズ | 家族を地域の情報資源、息抜き・支援活動へと導くため、ソーシャルワーカーが適切な形で関与する体制を確保する。 | 訪問看護の必要性を継続的に評価していく。 |
| 複数のサブスペシャルティ(訳注:より細かな専門分野に分けられた診療科をいう)の予約、機器、薬、物品を管理するケアコーディネーション | 障害者スポーツやスペシャル・オリンピックスへの参加を検討する。 |
- 治療の選択肢の詳細については、「遺伝性難聴・聴力喪失概説」のGeneReviewを参照
- 骨の基準点を把握すること、ならびに前庭神経や顔面神経の構造や走行状態(時に異常なことがある)を把握することは、手術計画を立てる上で重要である[Vesseurら2016c]。図5も併せて参照のこと。
- 場合によっては、顔面神経の走行に異常があるために、人工内耳手術が禁忌となるような例もある[Vesseurら2016a,Vesseurら2016c]。
図5:CHARGE症候群におけるCTとMRIの撮影
de Geusら[2017]より複製
(訳注:実質的には「表」であるが、原文では全体が写真になっているため、「図」となっている。)
| CHARGE症候群におけるCTとMRIの撮影 | |
| 目的 | CHARGE症候群において均質なMRI、CT撮影を行うこと。 CHARGE症候群患者において、気道に存在する難しい問題に気づくこと。 |
| 背景 | |
CHARGE症候群は、非常に広い臨床的スペクトラムを有する稀な先天奇形症候群である。
|
|
| 撮影部位 | |
CHARGE症候群罹患者については、次の撮影が必要になる。
必要に応じ:胸部のCT(喉頭、気管、気管支樹)、頸部のMRI、術前のナビゲーションのための後鼻孔のCT |
|
| CHARGE症候群の頭蓋撮影で確認されることのある典型的な異常 | |
|
|
| 推奨される撮影手法と撮影手順 | |
側頭骨のCT
脳のMRI
|
|
オランダ、フローニンゲン大学医療センター CHARGE症候群高度医療センター
発達遅滞/知的障害の管理に関する事項
以下に述べる内容は、アメリカにおける発達遅滞者、知的障害者の管理に関する一般的推奨事項を挙げたものである。
ただ、そうした標準的推奨事項は、国ごとに異なったものになることもあろう。
0-3歳
作業療法、理学療法、言語治療、摂食治療、乳幼児メンタルヘルスサービス、特別支援教育、感覚障害の治療が受けられるよう、早期介入プログラムへの紹介が望ましい。
これは、アメリカでは連邦政府が費用を負担して行われる制度で、すべての州で利用可能である。
個人個人の治療のニーズに合わせるため、在宅サービスの形で提供される。
3-5歳
アメリカでは、地域の公立学区(訳注:ここで言う「学区」というのは、地理的な範囲を指す言葉ではなく、教育行政単位を指す言葉である)を通じて発達保育園に入ることが推奨される。
入園前には、必要なサービスや治療の内容を決定するために必要な評価が行われ、その上で、運動・言語・社会・認知機能の遅れをもとに認定を受けた子どもに対して、個別の教育計画(IEP)が策定される。
こうした移行については、通常、早期介入プログラムが支援に当たる。
発達保育園は、基本的には通園を原則とするものの、医学的見地から通園が難しいと判断された子どもに対しては、在宅でサービスが提供されることもある。
全年齢
各地域、州、教育機関が適切な形で関与できるよう、そして、良好な生活の質を最大限確保する支援を親に対してできるよう、発達小児科医とよく話をすることが推奨される。
押さえておくべき事項がいくつかある。
- IEPのサービス
- IEPは、認定を受けた子どもに対して、特別に構成された指導と、それに関連するサービスを提供する。
- IEPのサービスは、必要な変更点を決定するため、毎年見直しが行われる。
- 特別支援教育に関する法律の定めに従って、子どもは学校という場で実現可能な範囲で最も制約の少ない環境に置かれる。
- 視覚や聴覚に関する相談員が子どものIEPのチームの一員として加わり、子どもが教材に接することができるよう支援を行う。
- IEPにおいては、子どもが教材に接するために必要と思われる範囲内で、理学療法、作業療法、言語治療の提供が行われる。それを超える部分については、罹患児のニーズに基づいて、自費での支援治療が検討されるようなこともある。行う治療の種類については、発達小児科医が個別に推奨を行う。
- 子どもがティーン世代になった時点で、移行計画についての話し合いが行われ、IEPの中にそれが組み込まれる。
IEPサービスを受ける人たちのため、公立学区はその人が21歳になるまでサービスを提供しなければならないことになっている。
そして、適切と判断された範囲内で、可能な限り多くの一般教育が受けられる環境が与えられる。
- 教室内で前のほうへの着席、補助機器の使用、代書者の起用、業間の休み時間の延長、宿題の変更、大きい教科書の使用といった配慮や変更が必要な人に対しては、504プラン(第504項:障害に基づく差別を禁じたアメリカの連邦法)が検討される。
- 発達障害者福祉局(DDA)への登録が推奨される。
- 収入が少なく、情報も不足している家族については、障害児のための補足的所得補償制度(SSI)の認定を受ける道がある。
- 州の盲聾者サービス:
- 上記のような全国レベルの教育サービスに加え、連邦政府が資金を拠出した州レベルの制度により、聴覚・視覚にまたがる問題を抱える出生から21歳までの人に対し、サービスが提供されることになっている(nationaldb.org/)。
- 州の盲聾者サービスの内容は、通常、家族に対して情報や訓練を提供すること、学校や早期介入プログラムに対して技術的な支援を行うこと、ならびに、IEPや移行に関する支援を行うことである。
DDAは、アメリカの公的機関で、認定を受けた人に対してサービスや支援を提供している。
認定基準は州によって異なるものの、ふつうは、診断名やそれに伴う認知/適応障害の度合に従って決定がなされる。
ここで注意が必要なのは、ここで言う「盲聾者」という言葉の意味が、視覚と聴覚の両方に問題があって、このサービスを受ける対象者を認定するための言葉であって、必ずしも聴覚ないし視覚の完全喪失を意味するのではないということである。
コミュニケーション
視覚障害を併せもつ場合でもそうでない場合でも、聴覚障害を有する子どもに対して生後6ヵ月未満の段階で聴覚リハビリテーションを開始すれば、正常な言語発達が得られることを示すデータが大きく積み上がってきている。
(その管理法に関する詳細については、「遺伝性難聴・聴力喪失概説」のGeneReviewを参照されたい。)
視覚・聴覚の障害の程度にもよるが、最初のコミュニケーションはタッチ・キュー(訳注:身振りや触り方での合図をいう)から始め、次いでオブジェクト・キュー(訳注:特定の物体を特定の活動の合図に用いることをいう)へ、そしてさらに聴覚言語(口からの言語)、ないし手話へと移行する。
こうした形で3歳までにコミュニケーションの訓練を開始することは、最終的に符合を用いたコミュニケーション(訳注:代表的には文字)を発達させる上で決定的に重要である[Thelin & Fussner 2005]。
利用可能なすべてのコミュニケーション手段(視覚・口腔感覚・触覚・身振り、ならびに手話や音声言語)を、早期から継続的に用いることが推奨されている。
たとえ最初は手話や体全体を使ったコミュニケーションから始めたとしても、年齢が上がるに従って、大多数の患児が口によるコミュニケーションへと移行できるようになる。
定期的追跡評価
表7:CHD7疾患罹患者について推奨される定期的追跡評価
| 系/懸念事項 | 評価の内容 | 実施頻度 |
|---|---|---|
| 体格 | 成長障害や肥満のチェックのための身長、体重測定。 | 来院ごと |
| 眼 | 矯正視力の悪化、白内障、網膜剥離(脈絡膜網膜コロボーマのみられる例)に関する眼科的診査 | 6ヵ月ごと、もしくは臨床的必要性に応じて |
| 機能的な視覚の状態をチェックするための聾-盲を専門とする医療者による評価 | 臨床的必要性に応じて | |
| 耳/鼻/喉 | 難聴の種類と程度、ならびに聴覚療育の成果を把握するための聴覚評価 | 年に1度、もしくは臨床的必要性に応じて |
| 耳の慢性感染症や浸出液に関する評価 | 思春期の間は来院ごと | |
| 口腔 | 歯のチェック | 3歳より後は少なくとも6ヵ月ごと |
| 心血管 | 不整脈が疑われるときは心電図とホルターモニター(特に複雑心奇形を有する罹患者) | 小児期後期/思春期初期から始め、臨床的必要性に応じて |
| 血圧測定 | 来院ごと | |
| 起立性頻拍症候群の兆候・症候に関する臨床的チェック | 思春期の人と成人について来院ごと | |
| 呼吸器 | 睡眠障害や閉塞性睡眠時無呼吸に関する臨床的チェック1 | 来院ごと |
| 消化器/摂食 | 嚥下障害2、逆流、便秘、腹痛/腹性ミグレイン(訳注:片頭痛症候群としての腹痛)の兆候・症候のチェック | 来院ごと;思春期、成人期には新たな摂食の問題が生じることがあることに注意。 |
| 筋骨格 | 脊柱側彎に関する臨床的チェックとX線によるチェック。 | 成長終了まで(場合によっては20代初期まで)の来院ごと |
| 神経 | 癲癇の症候に関するチェック | 来院ごと |
| 発達 | 粗大運動技能、微細運動技能、コミュニケーション能力のチェック;学習上の問題を調べるための学校に関することのチェック | |
| 内分泌 | 低ゴナドトロピン性性腺機能低下に起因する思春期遅延ないし停止を調べるための第2次性徴チェック | 7歳〜17歳まで |
| 甲状腺機能低下のチェック | 症候に応じて | |
| DXA法による骨密度測定 | 思春期から始め、定期的に3 | |
| 精神/行動 | 自己規制、衝動の制御、不安、執着、強迫、怒りなど、行動全般にわたるチェック | 幼児期以降、来院ごと |
- 扁桃が残存している人に対する扁桃肥大の評価を含む。
- 仮に思春期や成人の罹患者であっても、嚥下に関する評価は、敷居を低くして積極的に行う。
- 特に、低ゴナドトロピン性性腺機能低下の人、ならびに普段、ホルモン補充療法を受けている人。
避けるべき薬剤/環境
麻酔
CHARGE症候群の罹患者については、麻酔に伴う気道の問題が多くみられる。
これは、後鼻孔閉鎖、口唇裂口蓋裂その他の上気道の構造的奇形、ならびに関連脳神経の異常などに起因するものである。
気管軟骨が軟弱で、気管が柔らかいことが、麻酔の潜在的リスクを増大させる結果になっている。
神経原性の原因で、嚥下と喉頭蓋の閉鎖との協調運動が障害されているため、特に繰り返し全身麻酔を行う場合には、術後経過に問題が生じる可能性がある[Blakeら2009]。
麻酔後に気道の合併症が生じるリスクが高いため、麻酔の回数は最小限に制限し、可能な限り手術を1回にまとめて行うようにすべきである[de Geusら2017]。
リスクを有する血縁者の評価
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
CHD7疾患は常染色体顕性遺伝疾患であるが、新生の病的バリアントによって引き起こされる例が一般的である。
家族構成員のリスク
発端者の両親
- CHD7疾患と診断される罹患者の大多数は、新生の病的バリアントによって生じた例である[Janssenら2012]。
- 稀ながら、病的バリアントのヘテロ接合を有する片親からの継承により生じたCHD7疾患の罹患者が存在する。
- 発端者の両親については、遺伝的状態を明らかにして信頼性のある再発リスクのカウンセリングに繋げるため、分子遺伝学的検査を行うことが推奨される。
- 発端者において検出されたCHD7の病的バリアントが両親のいずれにおいても検出されなかった場合は、以下のような可能性を考慮する必要がある。
- 発端者の有する病的バリアントが新生のものであるという可能性。
CHD7の病的バリアントをヘテロで有する親を調べると、CHD7疾患の症候を1つないし複数もっていることがある[Mitchellら1985,Lalaniら2006,Delahayeら2007,Bergmanら2011b]。
注:
(1)発端者のもつ病的バリアントが両親のDNA中に検出されない
(2)親子鑑定の検査で、生物学的な親子関係が確認されている
という2つの条件を満たしたとき、報告書には「新生」と記載される。
- 両親の体細胞/生殖細胞系列モザイクの病的バリアントを、発端者が継承したという可能性。
- CHD7の病的バリアントの体細胞モザイクが実際に報告されている[Jongmansら2008]。
- (注:親の白血球DNAを検査することで、体細胞モザイクの例を漏れなく検出できるというわけではない。)
発端者の同胞
発端者の同胞の有するリスクは、発端者の両親の遺伝的状態により変わってくる。
- 発端者の片親がCHD7の病的バリアントを有していた場合、発端者の同胞がこの病的バリアントを継承するリスクは50%である。
- 発端者において検出されたCHD7の病的バリアントがいずれの親の白血球DNAからも検出されなかった場合、発端者の同胞への再現リスクは、経験的に約1%-2%であろうと思われる。
- CHD7の病的バリアントを継承した同胞に出現する重症度については、発端者の重症度から予測できるという性質のものではない。
それは、親の生殖細胞系列モザイクの可能性が残っているからである[Jongmansら2008,Pauliら2009]。
(CHD7疾患においては、現れる臨床症候に大きなばらつきがみられる;「臨床的特徴」の項を参照。)
発端者の子
- CHD7疾患の罹患者の多くは生殖能力を有しない。
- CHD7疾患罹患者の子が病的バリアントを継承する可能性は50%である。
- CHD7の病的バリアントをヘテロで有する子に現れる疾患の重症度については、発端者の重症度から推し量ることができるわけではない。
(CHD7疾患の臨床症候には大きな幅がみられる;「臨床的特徴」の項を参照)
他の家族構成員
他の血族の有するリスクは、発端者の両親の遺伝子の状況により変わってくる。
片親がCHD7の病的バリアントを有していた場合、その血族もリスクをもつことになる。
関連する遺伝カウンセリング上の諸事項
子どもがCHARGE症候群と診断された家族に対する遺伝カウンセリングについての総説は、Hefner & Fassi[2017]を参照されたい。
家族計画
- 遺伝的リスクの確定、出生前/着床前遺伝子検査を受けるかどうかの話し合いといったことに最も適しているのは、妊娠前の時期である。
- 罹患者の両親や軽微な症候を有する若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子を儲ける上での選択肢についての説明を含む)を提供することが望ましい。
出生前検査ならびに着床前の遺伝子検査
家系内に存在するCHD7の病的バリアントが同定された場合は、出生前検査ならびに着床前遺伝子検査を行うことが可能となる。
出生前検査の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。
現在、多くの医療機関では、出生前検査を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- ssociation CHRGE Enfant Soleil
France
www.associationcharge.fr - CHARGE Family Support Group
London
United Kingdom
Phone:020 8265 3604
Email:si_howard@hotmail.com
www.chargesyndrome.org.uk
- CHARGE Information Pack
Sense
CHARGE syndrome
- CHARGE Syndrome Association of Australia
Australia
Phone:61 480 121 345
Email:admin@chargesyndrome.org.au
www.chargesyndrome.org.au
- CHRGE Syndrome e.V.
Germany
Phone:0049-(0)9104-826 345
Email:info@charge-syndrom.de
www.charge-syndrom.de
- CHARGE Syndrome Foundation
18 Half Day Road #305
Buffalo Grove IL 60089
Phone:800-442-7604 (toll-free);516-684-4720
Fax:516-883-9060
Email:info@chargesyndrome.org
www.chargesyndrom.org
- My46 Trait Profile
CHARGE syndrome
- Perkins School for the Blind
e-learning videos on CHARGE syndrome
- Face Equality International
United Kingdom
Email:info@faceequalityinternational.org
www.faceequalityinternational.org
- National Consortium on Deaf-Blindness(NCDB)
Bibliography and links to state deafblind project resources
345 North Monmouth Avenue
Monmouth OR 97361
Phone:800-438-9376(toll-free);800-854-7013(TTY)
Fax:503-838-815
Email:info@nationaldb.org
www.nationaldb.org
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:CHD7疾患:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| CHD7 | 8q12.2 | クロモドメイン-ヘリカーゼDNA結合タンパク質7 | CHD7 database | CHD7 | CHD7 |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:CHD7疾患関連のOMIMエントリー(閲覧はすべてOMIMへ)
| 214800 | CHARGE SYNDROME |
| 608892 | CHROMODOMAIN HELICASE DNA-BINDING PROTEIN 7;CHD7 |
| 612370 | HYPOGONADOTROPIC HYPOGONADISM 5 WITH OR WITHOUT ANOSMIA;HH5 |
分子レベルでの病原
CHD7は、ATP依存性のクロマチンリモデリングに関与する1つのクロモドメインタンパク質をコードしている。
CHD7は、哺乳類のゲノム内で10,000以上の部位と結合し、他の何十もの遺伝子と相互作用を行う。
CHD7関連疾患にみられる症候は、リボソームにおける生合成の障害その他に、その原因があるように思われる。
CHD7は、複数のタンパク質から成る複合体の中で機能を発揮し、ATPのエネルギーを利用してヌクレオソームのリモデリングを行っている。
CHD7は、クロマチンの開閉度合の調節に関与し、「開」の時にはタンパク質が近づきやすく、「閉」の時にはタンパク質が近づきにくくなるといった形で、遺伝子発現の活性化、不活性化の調節を行っている。
その意味で、CHD7は一種のエピジェネティックな制御因子であると考えられている。
CHD7疾患の罹患者でみられる表現型の大きな多様性は、今もって完全には解明されていないものの、おそらく発生のさまざまな段階において、特定の細胞、特定の組織の中で、これが独自の役割を果たしているものと考えられる。
疾患を引き起こすメカニズム
CHD7が機能低下ないし機能喪失に至ることで、CHD7疾患の臨床症候を引き起こしている。
更新履歴:
-
Gene Reviews著者: Conny M van Ravenswaaij-Arts, MD, PhD, Meg Hefner, MS, Kim Blake, MD, MRCP, FRCPC, and Donna M Martin, MD, PhD
日本語訳者: 佐藤康守(たい矯正歯科)、水上都(札幌医科大学医学部遺伝医学) [ in present]
![]()