Joubert症候群
(Joubert Syndrome)
[Synonyms:JBTS, Joubert Syndrome and Related Disorders (JSRD), JS]
Gene Reviews著者: Melissa Parisi, MD, PhD and Ian Glass, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日:2017.6.29. 日本語訳最終更新日: 2023.4.2.
原文: Joubert Syndrome
要約
疾患の特徴
古典的Joubert症候群(JS)の大きな特徴は次の3つである。
- Molar tooth sign(MTS)と呼ばれる特徴的な小脳・脳幹の奇形
- 筋緊張低下
- 発達遅滞
こうした所見に加え、しばしば、反復頻呼吸ないし非定型眼球運動がみられる。一般に、年齢とともに呼吸の異常は改善していく一方で、体幹運動失調は時とともに進行していく。そして、粗大運動の発達指標の獲得は遅延する。認知機能の状態は、重度の知的障害から正常まで、幅がみられる。それ以外にも、網膜ジストロフィー、腎疾患、眼のコロボーマ、後頭部の脳瘤、肝線維症、多指趾、口腔の過誤腫、内分泌異常といったものがみられることがある。家系内においても家系間においても、大きなばらつきの幅がみられる。
診断・検査
JSの臨床診断は、特徴的臨床症候とMRI所見の2つをもとに行われる。現在までに、JSの原因遺伝子として34の遺伝子が知られている。うち33が常染色体潜性遺伝で、1つがX連鎖性である。JSの臨床診断を受けた例のおおむね62%-92%については、33のJS関連常染色体潜性遺伝子の1つの両アレル性病的バリアント、あるいは、つのJS関連X連鎖性遺伝子のヘテロ接合性病的バリアントの同定という形で、分子診断の確定が可能である。
臨床的マネジメント
症状に対する治療:
呼吸の異常を有する乳幼児・小児については、呼吸刺激剤(例えば、カフェイン)の使用、酸素補給、呼吸の機械的補助が必要になることがあり、稀には気管切開を要するようなこともある。これ以外の治療的介入としては、口腔運動機能障害に対する言語治療、作業療法や理学療法、教育的支援(例えば、視覚障害者に対する特別支援教育など)、胃瘻チューブによる栄養補給といったものが考えられる。多指趾、ならびに症状を伴う眼瞼下垂・斜視については、手術が必要になる場合がある。ネフロン癆、末期腎不全、肝不全、肝線維症については、標準治療を行う。
定期的追跡評価:
成長、視覚、肝機能、腎機能については、年に1度の評価を行う。神経心理学的検査、発達検査についても定期的に行う。
避けるべき薬剤/環境:
腎障害を有する例については、非ステロイド系抗炎症薬などの腎毒性を有する薬剤の使用を避ける。肝障害を有する例については、肝毒性を有する薬剤を避ける。
遺伝カウンセリング
JSの遺伝形式は、大半が常染色体潜性である。OFD1の病的バリアントに起因するJSについては、X連鎖性の遺伝形式をとる。2遺伝子遺伝の報告もみられる。
常染色体潜性遺伝については、罹患者の同胞の有するリスクは、受胎の段階で、罹患者となるリスクが25%、無症状の保因者となるリスクが50%、罹患者でも保因者でもない可能性が25%である。家系内の罹患者に存在する病的バリアントが特定済みの場合は、リスクを有する血縁者に対する保因者診断、高リスクの妊娠に備えた出生前診断、着床前遺伝学的検査を行うことが可能である。JSについて高リスクであることがわかっている妊娠については、超音波検査や、これに胎児MRIを併用した検査が有効とされている。
診断
Joubert症候群(JS)の診断基準については、今もって変遷が続いているものの、molar tooth signという神経放射線所見が必須であるという1点が、大半の研究者の一致した意見である[Valenteら2008,Parisi 2009,Brancatiら2010]。
Joubert症候群の診断は、以下の3つの主要な基準の存在を基にして行われる。
- Molar tooth sign
中脳と橋の境界部(峡部)を通るMRIの体軸断面における、小脳虫部の低形成と脳幹の異常を示す像をいう[Mariaら1997,Mariaら1999b,Quislingら1999]。Molar tooth signは、異常に深い脚間窩、幅広の直線状で目立つ上小脳脚、虫部すなわち小脳正中部の低形成により構成される(図1A,1B)[Mariaら1999b]。通常の軸位断、前額断、矢状断に加え、高性能のMRIで、後頭蓋窩を通る中脳から橋までの薄い(3mm厚)軸位断を作成することが推奨される。
- 乳児期の筋緊張低下、ならびに後年における運動失調の発生
- 発達遅滞/知的障害
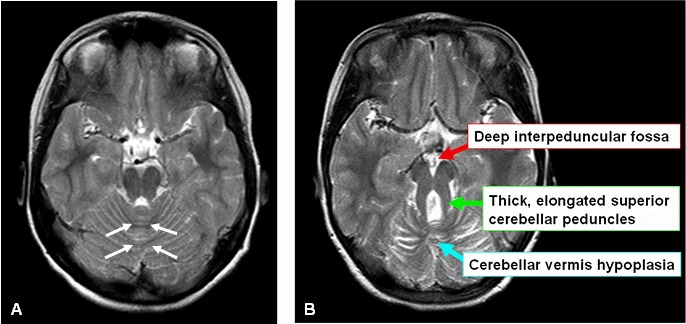
図1:Joubert症候群でみられるmolar tooth sign
A.健常者における小脳と脳幹を通る軸位断MRI画像。小脳虫部(白矢印で外形を示した)は正常である。
B.Joubert症候群の子どもの小脳と脳幹を通る軸位断MRI画像。Molar tooth signの3つの主要構成要素を矢印で示した。
JS罹患者においては、さらに次のような症候がみられることが多い。
- 呼吸パターンの異常(頻呼吸ないし無呼吸が交互に生じる)
- 眼球運動の異常 典型的には、眼球運動失行、あるいは、スムーズな追視ができず、注視・追視に際しての律動眼振といったことが生じる[Saraiva & Baraitser 1992,Steinlinら1997,Mariaら1999b,Tusa & Hove 1999]。
JS罹患者の半数未満にみられるその他の所見としては、網膜ジストロフィー、腎疾患、眼のコロボーマ、後頭部の脳瘤、肝線維症、多指趾、口腔の過誤腫、その他の異常がある。「その他」として挙げたこうした所見を有しない例については、従来、「古典型JS」あるいは「純粋型JS」という表現がなされてきた。しかし、実際には、もともと乳幼児期に古典型JSと診断された例の相当割合が、時の経過とともにこれらの所見を示すようになるという現実がある。
診断の確定
JSの臨床診断は、特徴的な臨床症候とMRI所見を有することをもって行われる。JSを引き起こすものとしては、現在までに34の遺伝子の病的バリアントが知られている。うち33が常染色体性、1つがX連鎖性である。33の常染色体潜性のJS関連遺伝子の両アレル性病的バリアント、あるいは、1つのX連鎖性のJS関連遺伝子のヘテロ接合性病的バリアントの同定といった形で、分子診断でのJSの確定が可能なのは、JSの臨床診断を受けた例の約62-94%である[Bachmann-Gagescuら2015a](表1aならびに1bを参照)。
分子遺伝学的検査のアプローチとしては、遺伝子標的型検査(マルチ遺伝子パネル)とゲノム検査(網羅的ゲノムシーケンシング)を組み合わせて用いるやり方が考えられる。遺伝子標的型検査の場合は、臨床医の側で疑わしい遺伝子の目星をつけておくことが必要となるが、ゲノム検査の場合、そうした必要はない。JSには大きな臨床的、遺伝的異質性が存在するため、Vilbouxら[2017]は、まずはマルチ遺伝子パネルから始め、そこで分子診断の確定に至らなかった場合はエクソームシーケンシングに進むといったやり方を提唱している。
- 34のJS関連遺伝子の一部ないし全部と、その他の関連遺伝子を含むマルチ遺伝子パネル(「遺伝子の上で関連のある疾患」の項を参照)
注:(1)パネルに含められる遺伝子の内容、ならびに個々の遺伝子について行う検査の診断上の感度については、検査機関によってばらつきがみられ、また、経時的に変更されていく可能性がある。
(2)マルチ遺伝子パネルによっては、このGeneReviewで取り上げている状況と無関係な遺伝子が含まれることがある。したがって、臨床医においては、どのマルチ遺伝子パネルを用いれば、現況の表現型と直接関係のない遺伝子の意義不明バリアントや病的バリアントの検出を抑えつつ、疾患の遺伝的原因の特定につながるかという点を事前に考えておくことが求められる。
(3)検査機関によっては、パネルの内容が、その機関の定めた定型のパネルであったり、表現型ごとに定めたものの中で臨床医の指定した遺伝子を含む定型のエクソーム解析であったりすることがある。
(4)ある1つのパネルに対して適用される手法には、配列解析、欠失/重複解析、ないしその他の非配列ベースの検査などがある。
本疾患に関しては、欠失/重複解析を含む検査とすることが推奨される(表1参照)。
マルチ遺伝子パネル検査の基礎的情報についてはここをクリック。
遺伝学的検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
- 網羅的ゲノム検査(臨床で利用可能な場合のみ)
網羅的ゲノム検査には、エクソームシーケンシングとゲノムシーケンシングがある。
網羅的ゲノム検査の基礎的情報についてはここをクリック。ゲノム検査をオーダーする臨床医に対する、より詳細な情報についてはここをクリック。
注:JSについては、臨床的・遺伝的異質性の幅がきわめて広いため、通常、単一遺伝子検査、あるいは直列型の単一遺伝子検査が有用になることはほとんどなく、したがって、こうしたものは推奨されない。ただ、以下のような民族的/祖先的背景を有する例については、場合によっては標的型の病的バリアントの解析を最初に行うことも考えられる。
- アシュケナージ系ユダヤ人:TMEM216におけるp.Arg73Leu[Edvardsonら2010]
- オランダ人:CPLANE1におけるp.Arg2904Ter[Kroesら2016]
- フランス系カナダ人:CPLANE1、CCD2A、NPHP1、TMEM231におけるいくつかのバリアント[Srourら2015]
- フッター派:TMEM237におけるp.Arg18Ter[Huangら2011]とCSPP1におけるc.363_364delTA[Shaheenら2014]
- 日本人:CEP290におけるc.6012-12T>A[Suzukiら2016]
JSの遺伝的原因として非常に多くみられるもの(すなわち、JS全体の中で、その遺伝子の病的バリアントの占める割合が1%以上に上るもの)については表1aを、JSの原因としてそれほど多くみられないもの(数家系のみで報告されている病的バリアント)については表1bを参照されたい。
表1a:分子遺伝学的にみたJoubert症候群:遺伝的原因として多くみられるもの
| 遺伝子1,2 | その遺伝子の病的バリアントがJS全体の中で占める割合 | その手法で病的バリアント3が検出される割合 | |
|---|---|---|---|
| 配列解析4 | 遺伝子標的型欠失/重複解析5 | ||
| AHI1 | 7%-10%近く6,7,8 | 95%超 | 脚注9参照 |
| CPLANE1 | 8%-14%7,8,10 | 100% | 報告例なし |
| CC2D2A | 8%-11%近く7,8,11 | 100%近く | 脚注12参照 |
| CEP290 | 7%-10%7,8,13,14 | 99%近く | 脚注15参照 |
| CSPP1 | 2%-4%7,8,16 | 100% | 報告例なし |
| INPP5E | 2%-4%7,8 | 100% | 報告例なし |
| KIAA0586 | 2%-7%近く8,17 | 2例の報告があり、うち1人はマルチエクソンの欠失18 | |
| MKS1 | 2%-6%近く7,8,19 | 95%近く | 脚注20参照 |
| NPHP1 | 1%-2%近く7,8,21,22 | 脚注22参照 | 20%-25% |
| RPGRIP1L | 1%-4%7,8,23 | 100% | 報告例なし |
| TCTN2 | 1%近く7 | 13人中13人24 | 報告例なし |
| TMEM67 | 6%-20%近く7,8,9,12,25 | 99%近く | 脚注26参照 |
| TMEM216 | 2%-3%近く7,8,27 | 8人中8人26 | 報告例なし |
本表に挙げた遺伝子の病的バリアントは、いずれもJS全体の中で1%超を占める。
- 遺伝子の配列はアルファベット順。
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- その遺伝子で検出されている病的バリアントの情報については、「分子遺伝学」の項を参照のこと。
- 配列解析を行うことで、benign、likely benign、意義不明、likely pathogenic、pathogenicといったバリアントが検出される。バリアントの種類としては、遺伝子内の小さな欠失/挿入、ミスセンス・ナンセンス・スプライス部位バリアントなどがあるが、通常、エクソン単位あるいは遺伝子全体の欠失/重複については検出されない。
- 遺伝子標的型欠失/重複解析では、遺伝子内の欠失や重複が検出される。具体的手法としては、定量的PCR、ロングレンジPCR、MLPA法、あるいは単一エクソンの欠失/重複の検出を目的に設計された遺伝子標的型マイクロアレイなど、さまざまなものがある。
- Parisiら[2006],Valenteら[2006a]
- Bachmann-Gagescuら[2015a]は、375家系、440人の罹患者について、27のJS関連遺伝子の病的バリアントを調べている。
- Vilbouxら[2017]は、86家系、100人について27の遺伝子を含むマルチ遺伝子パネルとエクソームシーケンシングを用いて調べ、うち81人(94%)で病的バリアントを同定している。
- 9.3つの報告がみられる[Utschら2006,Bachmann-Gagescuら2015a,Watsonら2016]。
- Kroesら[2016]は、JSを有する51人の北欧人コホートで、22のJS関連遺伝子と599の繊毛関連遺伝子の評価を行っている。その他のコホートと違って、このコホートでのCPLANE1の病的バリアントの割合は12%に及んでいる。
- Gordenら[2008],Dohertyら[2010]。1つの大コホートにおけるCC2D2Aの病的バリアントの出現頻度は、209人中16人(7.7%)であった[Bachmann-Gagescuら2012]。
- 12.2つの報告例が存在する[Mougou-Zerelliら2009,Suら2015]。
- Sayerら[2006],Valenteら[2006b],Valenteら[2008],Travagliniら[2009],Bachmann-Gagescuら[2015a]のデータからは、7%-10%という数字が支持される。一方、北欧人コホートで、51人中1人(2%)にしかみられなかったとするものもある[Kroesら2016]。
- Suzukiら[2016]は、30家系のコホート(3人を除く全員が日本人)中の83%に病的バリアントが検出され、その内容は、TMEM67(コホートの26%)、CEP290(コホートの22%)、OFD1、INPP5E、AHI1、CPLANE1(それぞれコホートの7.4%)であったと報告している。
- 15.1例が報告されている[Travagliniら2009]。
- Tuzら[2014],Akizuら[2014]
- ある1つのJSコホートにおいて、KIAA0586の病的バリアントは366家系中9家系(2.5%)を占めていた。 ただ、c.428delGの1塩基欠失が一般集団内で高頻度にみられる[Roosingら2015]こと、ならびに、その臨床的表現型が大きな幅をもつ[Albyら2015,Malicdanら2015]ことから考えて、KIAA0586の病的バリアントは、実際はもっと多く存在する可能性がありうるように思われる。
- KIAA0586の複合ヘテロ接合性病的バリアントを示した6例中3例については、一方のアレルの病的バリアントがエクソン8-10にまたがる800bpの欠失であった[Malicdanら2015]。
- MKS1の病的変化を報告したものは2つで、そのうちの1つ[Romaniら2014]では260人のJS罹患者中2人、他方[Slaatsら2016]では371のJS家系中9家系でこれがみられたという。
- 20.4つの報告がみられる[Kyttäläら2006,Frankら2007,Abu-Safiehら2012,Szymanskaら2012]。
- ネフロン癆を有する罹患者についていうと、もう少し値が高くなる可能性がある。
- 22.稀ながら、JS罹患者の中にホモ接合の形での欠失を有する例がみられる。 欠失/重複解析単独の場合は、欠失がヘテロで生じている状態は検出可能であるがNPHP1の1塩基バリアントについては検出されない。このホモ接合の遺伝型は稀であるように思われる。検出例が最も多いのは、290kb近くのサイズの頻出欠失(common deletion)である。
- Artsら[2007],Delousら[2007],Parisi [2009]
- Juric-Sekharら[2012],Bachmann-Gagescuら[2015a]
- Baalaら[2007],Brancatiら[2009],Dohertyら[2010]
- 26.1つの報告例がみられる[Khaddourら2007]。
- JSの462家系中14家系(3%近く)でTMEM216の病的バリアントが確認されている[Valenteら2010]。
- Valenteら[2010],Leeら[2012b](訳注:表中に「28」の数字が記載された箇所はない。「TMEM216」の欄にある「26」が「28」の誤りであるように思われる。)
配列解析の結果の解釈に際して留意すべき事項についてはこちらをクリック。
表1b:分子遺伝学的にみたJoubert症候群:遺伝的原因として比較的少ないもの
| 遺伝子1,2,3 | コメント |
|---|---|
| ARL13B | 2家系;表現型は、古典型JSから、後頭部脳瘤と色素性網膜症を伴うJSまで幅がみられる[Cantagrelら2008];欠失/重複の報告はみられない。 |
| B9D1 | 2家系でみられ、両家系とも「純粋型」JS;本遺伝子に生じる病的バリアントはMeckel症候群(MKS)の原因ともなっている。欠失/重複の報告はみられない[Romaniら2014]。 |
| B9D2 | 2家系でみられ、多指趾は両家系ともにみられ、脳瘤・口蓋裂・舌の過誤腫は1家系のみでみられている;本遺伝子に生じる病的バリアントはMeckel症候群の原因ともなっている。欠失/重複の報告はみられない[Bachmann-Gagescuら2015a]。 |
| C2CD3 | 1報告中の2家系で同定されており、両家系とも、口蓋裂ないし口腔の過剰小帯を呈しており、口-顔-指症候群を思わせるものとなっている。欠失/重複の報告はみられない[Bachmann-Gagescuら2015a]。 |
| CEP41 | 少なくとも725人のJS罹患者をチェックし、既知のJS関連遺伝子の病的バリアントを有する多くの例を除外した残りの中で、3家系8人に、CEP41の病的バリアントがみられたことが報告されている。 50%をわずかに超える数の罹患者に、片側性あるいは両側性の軸後性多指趾がみられている。網膜疾患のみられた罹患者は2人のみで、そのうちの1人は片側性コロボーマ、片側性腎疾患、判別不明性器を有し、生後7日で死亡している。1家系では、男性罹患者全員に小陰茎がみられ、2人に成長ホルモン分泌不全がみられた。スプライス部位バリアントのみが報告されており、欠失/重複の報告はみられない[Leeら2012a]。 |
| CEP104 | 3家系の報告があり、すべて「純粋型」JS;欠失/重複の報告はみられない[Srourら2015] |
| CEP120 | JS罹患者491人中4人がこの遺伝子のミスセンス・フレームシフト・ナンセンス・スプライス部位バリアントであった;表現型は「純粋型」JSからMeckel症候群、口-顔-指症候群、Jeune呼吸不全性胸郭異形成症に至るまで幅がみられた;欠失/重複の報告はみられない[Shaheenら2015b,Roosingら2016a]。 |
| IFT172 | JS罹患者440人中1人がこの遺伝子の病的ミスセンスバリアントであった[Bachmann-Gagescuら2015a]。ミスセンス型ないしトランケーション型病的バリアントを有していた12家系中2家系は、網膜ジストロフィー、肝線維症、ネフロン癆、小脳虫部低形成など、JSの症候とJeune呼吸不全性胸郭異形成症の症候(1家系についてはMainzer-Saldino症候群の症候も)が重なった症候を示した。欠失・重複の報告はみられない[Halbritterら2013]。 |
| KATNIP (KIAA0556) |
血族結婚の1家系の3同胞に、この遺伝子のトランケーション型病的バリアントのホモ接合が確認されており、3人中2人が汎下垂体機能低下(男性のほうは小陰茎、女性のほうはMRIでの下垂体低形成)を示した[Sandersら2015]。これとは別のやはり血族結婚の1家系でも、古典型JSの症候を有する2同胞にトランケーション型病的バリアントのホモ接合が確認されている;欠失・重複の報告はみられない[Roosingら2016b]。 |
| KIF7 | 440家系中3家系で、この遺伝子の病的バリアントが同定されている[Bachmann-Gagescuら2015a]。口-顔-指症候群の症候が多くみられ、これに脳梁無形成/低形成、水頭症、大頭症といった中枢神経系の所見が伴う場合がある[Dafingerら2011,Putouxら2011]。多指趾とこうした中枢神経系所見の組合せは、先端脳梁症候群ないしhydrolethalus症候群を思わせる[Putouxら2011]。ナンセンスバリアントとフレームシフトバリアントが主体;欠失・重複の報告はみられない。 |
| OFD1 | X連鎖;欠失・重複の報告はみられない。440家系中4家系[Bachmann-Gagescuら2015a]、250家系中2家系(罹患者が男性のみの84家系中2家系)[Coeneら2009]で、この遺伝子の病的バリアントが確認されている。脳瘤、水頭症、大頭症、多小脳回、多指趾、網膜疾患などがその症候である。嚢胞性腎疾患、水頭症、大頭症、多小脳回を有する1家系も報告されている[Fieldら2012]。 |
| PDE6D | 1家系の3同胞(スプライス部位バリアントのホモ接合)が報告されており、腎発育不全、網膜ジストロフィー、小眼球、眼のコロボーマ、軸後性多指趾などがその表現型である[Thomasら2014]。 |
| POC1B | Leber先天黒内障、腎肥大、多発性嚢胞腎(ネフロン癆より常染色体顕性多発性嚢胞腎に似る)に加えて、肝線維症を伴わないJSの典型症候を有するイラク人大家系において、この遺伝子の病的ミスセンスバリアントのホモ接合が同定されている。注目すべきことに、重度で緩徐進行性の錐体杆体ジストロフィーのみでJSの症候を伴わない1家系においても、これと同一の病的バリアントのホモ接合が同定されている[Beckら2014]。欠失・重複の報告はみられない。 |
| TCTN1 | 440家系中1家系で、この遺伝子の病的バリアントが確認されている[Bachmann-Gagescuら2015a]。ホモ接合性のスプライス部位バリアントを有する2同胞は、前頭・側頭葉の脳回肥大を示したものの、網膜や腎の所見は示さなかった[Garcia-Gonzaloら2011]。欠失・重複の報告はみられない。 |
| TCTN3 | 440家系中1家系で、この遺伝子の病的バリアントが確認されている[Bachmann-Gagescuら2015a]。また、既知のJS遺伝子を有する家系を除いた58家系中1家系に、この遺伝子の両アレル性病的バリアントを確認したとする報告もみられる[Thomasら2012]。重度の、出生前致死型口-顔-指症候群(OFD)Ⅳ型(Mohr-Majewski症候群;MJS)を有する5家系において、この遺伝子のトランケーションバリアントのホモ接合が同定されているものの、その表現型の中に、軸後性多指趾、嚢胞性腎疾患、胆管増生、後頭部脳瘤といったものが含まれていることから考えて、実際にこれがOFDの1つの型、すなわちMeckel症候群であるか否かは議論の余地のあるところである。この遺伝子のホモ接合性ミスセンスバリアントを有するトルコ人1家系の2発端者は、ばらつきの幅をもった軸後性多指趾を伴う脊柱側彎、口腔所見、馬蹄腎、心室中隔欠損を示した[Thomasら2012]。欠失・重複の報告はみられない。 |
| TMEM107 | JSあるいは口-顔-指症候群(OFD)Ⅳ型を有する罹患者238人中、血族結婚で生まれた双生児1組が、この遺伝子のミスセンスバリアントのホモ接合を有し、網膜症、ならびに術後性多指趾を含むOFDの症候を呈した;そして、古典型JSと網膜症を呈した別の1男性が、この遺伝子の病的バリアントを複合ヘテロで有していた[Lambacherら2016]。欠失・重複の報告はみられない。 |
| TMEM138 | 440家系中1家系で、この遺伝子の病的バリアントが確認されている[Bachmann-Gagescuら2015a]。血族結婚のアラブ人8家系11人では、コロボーマ(6人)、網膜ジストロフィー(3人)、嚢胞腎もしくはネフロン癆(3人)がみられた。多指趾の報告もみられ、Meckel症候群の1胎児では脳瘤がみられた[Leeら2012b]。欠失・重複の報告はみられない。 |
| TMEM231 | フランス系カナダ人JS罹患者の一部に、この遺伝子の病的バリアントがみられる。2家系の3人は重度の表現型(歩行不能,攻撃的行動,自立生活技能の欠如)を示した。2人には肉眼所見としての腎嚢胞、ならびに網膜疾患、1例には軸後性多合指趾がみられた[Srourら2012a]。この遺伝子と、その偽遺伝子との間での遺伝子変換イベントの報告がみられる[Maglicら2016]。 |
| TMEM237 | 440家系中1家系で、この遺伝子の病的バリアントが確認されている[Bachmann-Gagescuら2015a]。JS罹患者では201人中2人のみ、そしてMeckel症候群/JS罹患者の90人で、この遺伝子の病的バリアントが確認されている[Huangら2011]。このタイプのJSは、当初、フッター派の集団内でみられたMeckel症候群として報告されていたものである[Boycottら2007]。なお、フッター派内における保因者頻度は6%と推定されている[Huangら2011]。脳瘤、水頭症、嚢胞性腎疾患が多くみられる。両アレル性病的バリアントを有するオーストリアの1大家系では、「朝顔状視神経乳頭奇形」も報告されている[Janeckeら2004,Huangら2011]。TMEM237のエクソン1と1aから隣接遺伝子にまでまたがる24kbの欠失が確認されている[Watsonら2016]。 |
| TTC21B | 現在のところ、この遺伝子に両アレル性の病的バリアントを有するJS罹患者は報告されていない。片アレル(ヘテロ接合性)の病的バリアントのもつ機能面での意義はよくわかっていない。ヘテロ接合の3人については、その臨床的状態に関する報告はなされていない。「TTC21B」の「病的バリアント」(本稿末尾)の項を参照(訳注:本編の後ろに訳出している)。繊毛病を有する臨床的に多様な753人のコホート中、5%にこの遺伝子の病的バリアントが確認されているが、そのうち別の繊毛病関連遺伝子にも併せて病的バリアントがみられた例は33%に過ぎなかった[Davisら2011]。 |
| ZNF423 | 乳児期発症のネフロン癆、脳梁低形成、内臓逆位を呈する血族結婚の1家系で、この遺伝子のホモ接合性病的バリアントが確認されており、それ以外のJS罹患者96人中2人に、ドミナントネガティブ作用による機能喪失に至るとされる(ただし、証明されているわけではない)この遺伝子の特定の相互作用ドメインの変化のヘテロ接合がみられた[Chakiら2012]。欠失・重複の報告はみられない。 |
- 遺伝子の配列はアルファベット順。
- 染色体上の座位ならびにタンパク質に関しては、表A「遺伝子とデータベース」を参照。
- これらの遺伝子については、「分子遺伝学」の項で詳述していない。
一部については、別に本稿末尾で述べている(訳注:本編の後ろに訳出している)。
臨床的特徴
臨床像
古典型Joubert症候群は、molar tooth sign(MTS)と呼ばれる特徴的小脳・脳幹奇形、筋緊張低下、発達遅滞という3主徴を特徴とする。これらに加え、時折生じる頻呼吸や無呼吸、非定型的眼球運動といったものを伴うことも多い。一般に、呼吸の問題が年齢とともに改善していく一方で、経時的に体幹運動失調が現れ、粗大運動発達指標の達成が遅延する。
認知機能は、重度の知的障害から正常まで、ばらつきの幅が大きい。
その他、現れる可能性のある所見としては、網膜ジストロフィー、腎疾患、眼のコロボーマ、後頭部の脳瘤、肝線維症、多指趾、口腔の過誤腫、内分泌異常といったものがある。表2は、表現型ごとにみた表現型と遺伝子の関連、表3は、遺伝子ごとにみた表現型と遺伝子の関連を示したものである。JSにおいては、家系内・家系間とも、表現型のばらつきがみられる。
JSの臨床的特徴の多くは、乳児期に明らかになる[Joubertら1969,Boltshauser & Isler 1977]。眼振、眼球運動失行、呼吸パターンの異常といったものは、JSのサブタイプのいずれにおいても現れる可能性がある。JSの子どもの大多数は、体幹運動失調をきたし、筋緊張低下とあいまって、粗大運動発達指標の達成が遅延する。
眼振
Joubert症候群の子どもの大多数については、出生時に水平性眼振がみられるものの、これは年齢が進むとともに改善していく。また、回旋眼振や振子眼振の報告もみられる。
眼球運動失行
眼球運動失行は、乳児期よりも小児期に発見されることが多いが、これはおそらく、その時期まで見逃してしまうことが多いためであるように思われる[Steinlinら1997]。眼球運動失行を有する子どもの多くは、衝動性眼球運動(サッカード;saccade)ができないため、その代償行動としての衝動性頭部運動を示す[Hodgkinsら2004,Khanら2008,Weissら2009]。水平性の頭部揺動(すなわち、いやいやをするような頭の振戦)が2歳未満の乳幼児で報告されている[Porettiら2014]。出生時に明らかに異常な眼球運動がみられる場合でも、年齢とともに成人型視覚に移行して、視力やfunctional visionは改善していくことがある[M ParisiとA Weissの個人的見解]。
呼吸所見
JS罹患児の多くは、新生児期を中心に、無呼吸、頻呼吸、あるいは、その2つが時として交互に現れるといった異常を示す[Saraiva & Baraitser 1992,Steinlinら1997,Mariaら1999a,Valenteら2008]。一部、無呼吸で死亡に至る例がみられるものの、時折生じていた無呼吸が年齢とともに改善し、完全に消失するような例もみられる[Mariaら1999b]。
JSの子どもは、中枢性(特に乳児期から小児期にかけて)、閉塞性(特に、舌肥大、筋緊張低下、肥満が関係してくる学童期から思春期にかけて)の両方の睡眠時無呼吸に関し、高リスク状態にある[Parisiら2009]。妥当性検証済の睡眠質問票を用いたJS罹患者に対する自己評価による睡眠行動調査では、調査した14人中6人に、睡眠関連呼吸障害の存在が示唆されている[Kamdarら2011]。CEP290の両アレル性病的バリアントによって生じるLeber先天黒内障罹患者の中に、運動型繊毛の異常のため、慢性鼻炎、反復性副鼻腔炎、気管支炎といった呼吸器症状を起こしやすい例がみられることが報告されている[Paponら2010]。
中枢神経系の所見
- 認知機能
認知機能は、重度の知的障害から正常な認知機能を有するものまで多様で[Porettiら2009]、大学で学ぶような罹患者も数例みられる。知的障害がみられる例について言うと、その程度は中等度であることが多い[Steinlinら1997,Hodgkinsら2004,Bulgheroniら2016,Summersら2017]。JS罹患者110人を用いた研究で、小脳虫部低形成の程度と高次脳機能障害との間に相関がみられることが明らかになっている[Porettiら2017]。
- 発語失行
これは多くみられる所見であり、スピーチの理解と発話能力との乖離は、この発語失行という言葉で説明しうるものであるように思われる[Hodgkinsら2004,Braddockら2006]。
- 脳波の異常ないし癲癇発作
一部の罹患者で脳波の異常や癲癇発作がみられるものの、正確な発生率は不明である[Saraiva & Baraitser 1992]。脳波の異常を伴うJS罹患者ほど、高次脳機能障害の程度が重度であるとする1研究がみられる[Summersら2017]。
- 自閉症
一部のJS罹患児に自閉症が報告されている[Holroydら1991,Ozonoffら1999]ものの、より最近の調査によると、こうした行動障害の類の多くは、古典的自閉症スペクトラム障害ではないということが示唆されている[Takahashiら2005]。
- 行動上の問題
一部の子どもや思春期の罹患者に、不注意、多動、ならびに癇癪をはじめとする非定型行動といった行動上の問題がみられる[Deonna & Ziegler 1993,Hodgkinsら2004,Farmerら2006]。JS罹患者54人を対象としたある調査で、情緒面や行動上の問題が40%近くの罹患者にみられたと報告されている[Bulgheroniら2016]。76人を対象とした別の調査によると、行動上の問題は、外在化問題行動(攻撃性,反抗的挑戦)より内在化問題行動(不安,抑鬱)の形で現れやすいという[Summersら2017]。
JSの臨床的サブタイプ
表2ならびに表3を参照されたい。
表2:Joubert症候群:臨床的サブタイプ
| 臨床的サブタイプ名 | 一次基準1以外の必須症候 | 強く関連する症候2 | 別名 | 遺伝子(太字は主要遺伝子) |
|---|---|---|---|---|
| 古典型あるいは純粋型Joubert症候群 | JS JS type A |
多数の遺伝子 | ||
| 網膜疾患を伴うJoubert症候群(JS-Ret) | 網膜ジストロフィー(Leber先天黒内障を含む) | JS type B | AHI1 CEP290 CEP41 INPP5E MKS1 TMEM107 TMEM138 TMEM216 |
|
| 腎疾患を伴うJoubert症候群(JS-Ren) | ネフロン癆(嚢胞性腎疾患を含む) | AHI1 CC2D2A CEP290 NPHP1 OFD1 RPGRIP1L TMEM138 TMEM216 TMEM237 ZNF423 |
||
| 眼・腎疾患を伴うJoubert症候群(JS-OR) | 網膜ジストロフィー(Leber先天黒内障を含む) ネフロン癆 |
先天性肝線維症(時折) | JS type B CORS Senior-Løken症候群 Dekaban-Arima症候群 |
AHI1 CC2D2A CEP290 NPHP1 POC1B RPGRIP1L TMEM216 TMEM231 TMEM237 |
| 肝疾患を伴うJoubert症候群(JS-H) | 先天性肝線維症 | コロボーマ ネフロン癆 |
COACH症候群 Gentile症候群 |
CC2D2A CEP290 INPP5E RPGRIP1L TMEM67 |
| 口-顔-指症候を伴うJoubert症候群(JS-OFD) | 舌の過誤腫 口腔の小帯 多指趾3 |
口唇裂/口蓋裂 | Varadi-Papp症候群 口-顔-指症候群Ⅵ型 口-顔-指症候群Ⅳ型 Mohr-Majewski症候群 |
B9D2 C2CD3 CPLANE1 CEP120 KIF7 OFD1 TCTN2 TCTN3 TMEM107 TMEM216 |
| 先端脳梁症候を伴うJoubert症候群(JS-AC) | 骨格異形成(短肋骨,小さな胸郭,短肢,嚢胞性腎疾患) | 水頭症 | 先端脳梁症候群 | KIF7 |
| Jeune呼吸不全性胸郭異形成症候を伴うJoubert症候群(JS-JATD) | 多指趾3 円錐形の骨端 先天性肝線維症 |
Jeune呼吸不全性胸郭異形成症 Mainzer-Saldino症候群 |
CEP120 CSPP1 IFT172 KIAA0586 |
Brancatiら[2010]より引用。
症候の様相には極度に大きな臨床的異質性がみられ、また、ここに挙げた症候の多くは、発症年齢に大きなばらつきの幅がみられる。そのため、この分類の枠組みは確定的なものではないと理解されたい。
AC=先端脳梁;COACH=小脳虫部低形成(cerebellar vermis hypoplasia),精神発達遅滞(oligophrenia),運動失調(ataxia),コロボーマ(coloboma),肝線維症(hepatic fibrosis);CORS=小脳-眼-腎症候群(cerebello-oculo-renal syndrome);H=肝;JATD=Jeune呼吸不全性胸郭異形成症;OR=眼-腎;Ren=腎;Ret=網膜
- 一次基準=molar tooth sign(MTS),筋緊張低下,発達遅滞(DD)
- これらのサブタイプにおいては、その他に、脳瘤、軸後性多指趾、他の構造的脳奇形(多小脳回を含む)、先天性心疾患、Hirschsprung病、内臓逆位といった症候がみられることがあるものの、これらは大症候ではない。
- 多指趾は、特に手の場合は軸後性が多く、足については軸前性が多い。 OFDⅥに特徴的なのは、Y字形の中手骨を含む中央列の多指。
網膜疾患を伴うJoubert症候群(JS-Ret)
網膜疾患を伴うJoubert症候群は、色素性網膜症を特徴とし、これは時に典型的な網膜色素変性症と区別できないようなこともある。これは、時として、新生児期に発症する先天性盲、ならびにLeber先天黒内障(LCA)に類似した減弱型ないし消失型の網膜電図を示すほど重度の場合がある[Tusa & Hove 1999]。ただ、網膜疾患は非進行性の場合もあり、そしてまた、乳幼児期に必ずみられるといったものでもない[Steinlinら1997]。JSRD(訳注:このGeneReviewでは、古典型Joubert症候群を「JS」と略するのに対し、その関連疾患まで広く捉えたJoubert症候群を「JSRD」と略している)の235家系を対象としたある調査では、網膜ジストロフィーが30%に確認されている[Doherty 2009]。
- 眼のコロボーマ
眼のコロボーマは、脈絡膜網膜に最も多くみられ[Saraiva & Baraitser 1992,Parisiら2009]、COACH症候群のバリアントでみられるような肝線維症を伴うこともある[Dohertyら2010]。ある調査によると、JSRDを有する家系の19%にコロボーマがみられたという[Doherty 2009]。「朝顔状視神経乳頭奇形」と表現される網膜変化が、TMEM237の両アレル性病的バリアントを有するオーストリア、チロル地方の1大家系で報告されている[Janeckeら2004,Huangら2011]。
- その他
多様な形でみられる。
- 眼瞼下垂,斜視ないし弱視
- 第Ⅲ脳神経麻痺[Hodgkinsら2004]
腎疾患を伴うJoubert症候群(JR-Ren)
腎疾患を伴うJoubert症候群は、もともとは2つの型(ネフロン癆,嚢胞性腎異形成)として報告されてきた。しかし現在では、これら2つは1つの連続体を構成するものであって、腎疾患の段階に従って腎症候に異なった形で特定の変化が現れたものであろうと考えられている。慢性尿細管間質性腎症の1型である若年性ネフロン癆は、多くの場合、多飲、多尿、尿濃縮能障害、発育遅延、貧血といったものを伴って10歳未満あるいは10歳代で現れる。そして、平均で13歳には末期腎疾患にまで進行する[Hildebrandtら1998]。超音波検査で確認可能な腎の変化は、上記の経過の後半でみられるようになり、エコー輝度の上昇を伴う矮小の瘢痕化腎や、時として、嚢胞性腎異形成でみられる所見である皮髄境界部の嚢胞の形で現れる[Saraiva & Baraitser 1992,Steinlinら1997,Satranら1999]。
ネフロン癆と嚢胞性腎異形成が形作るスペクトラム以外に、常染色体潜性多発嚢胞腎症(ARPKD)類似の第2のタイプの腎疾患の報告もみられる。
- TMEM67の両アレル性病的バリアントに起因するJS罹患者3人に、ARPKDに特徴的な腎疾患、すなわち、腎肥大、瀰漫性小嚢胞腎、重度の早発性高血圧、先天性肝線維症がみられ、さらにはネフロン癆に特徴的な慢性貧血がみられたという報告がみられる[Gunay-Aygunら2009]。
- フッター派の集団において、TMEM237の病的バリアントに起因して生じたJSの発端者の約70%に嚢胞性腎疾患と腎機能異常がみられ、さらに、一部については、高血圧も報告されている[Boycottら2007,Huangら2011]。
腎疾患は、JS罹患者の23%[Doherty 2009]、30%[Saraiva & Baraitser 1992]にみられたと報告されている。腎疾患は小児期や思春期に現れることから、コホートの年齢が上がると、こうした発生頻度の値も上昇する可能性がある[Steinlinら1997]。
眼・腎疾患を伴うJoubert症候群(JS-OR)
腎疾患と網膜の異常が、時として1人の罹患者に同時に現れる。多くのJS関連遺伝子については、嚢胞性腎疾患と網膜ジストロフィーが同時に現れるが、この両者の組合せは、時として、Senior-Løken症候群と呼ばれるものである[Parisiら2009,Brancatiら2010](表2)。JS-ORは、過去にはDekaban-Arima症候群(網膜症,嚢胞性異形成腎)と呼ばれたこともある。これは、出生前あるいは出生時に明らかになる。
肝疾患を伴うJoubert症候群
肝線維症は、通常、進行性で、出生の段階ですでに症状があることは稀である。先天性肝線維症は、門脈胆道系の発生障害で、その特徴は、組織学的には、胆管板の形成異常(胆管板奇形)、肝門脈の分枝異常、門脈域の進行性線維症である。臨床所見としては、肝臓の肥大と形態異常、比較的よく保存された肝細胞機能、そして、脾腫・脾機能亢進・胃食道静脈瘤を引き起こす門脈圧亢進などがある。
あるコホートでは、JS罹患者の18%に肝線維症がみられている[Doherty 2009]。
JSでこれがみられる場合は、しばしば脈絡網膜コロボーマ、そして時に腎疾患が伴う。コロボーマ、高次脳機能障害(精神発達遅滞)、運動失調、小脳虫部低形成、肝線維症の組合せは、COACH症候群と呼ばれる[Satranら1999,Gleesonら2004,Dohertyら2010]。
口-顔-指症候を伴うJoubert症候群(JS-OFD)
口腔症状としては、正中上口唇裂、舌正中の溝形成、歯槽頂縁の過誤腫(図2B)、口蓋裂、口腔の小帯形成、舌の分葉ないし過誤腫などがある。口腔顔面症候としては、眼間開離もしくは内眼角開離、鼻翼低形成、小下顎などがある。
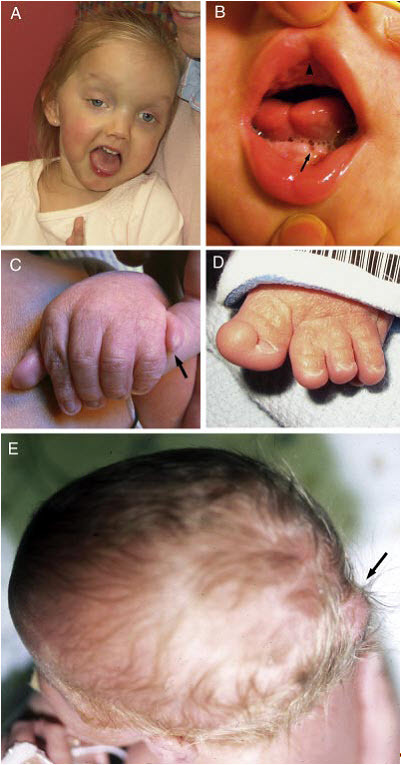
図2:JSRDの臨床症候
A.27ヵ月のJS/COACH症候群女児の顔面症候。幅広の額、高い眉毛、斜視、眼瞼下垂(右眼)、顔面の緊張低下を意味する締まりのない口などがみられる。
B.口-顔-指症候群様症候を有するJSの子どもの口腔内所見。正中上唇裂(▲)、舌正中の溝形成、下顎歯槽頂縁の凹凸(➡)がみられる。
C.軸後性多指を有するJS乳児の左手(➡)。
D.拇趾の軸前性多趾を有するJS乳児の左足。
E.頭蓋骨後頭部の突出を伴う後頭部の小さな脳瘤を有する乳児の上面観(➡)。
Parisiら[2009]より許可を得て転載。
顔写真の使用に関しては、家族の許可も併せて得ている。
多指趾は、発端者の8%-19%にみられる[Doherty 2009,Brancatiら2010]。多指趾は片側性のこともあれば、両側性のこともあり、多くが軸後性である(図2C)が、足については軸前性多趾もしばしば報告されている[Saraiva & Baraitser 1992]。
JS罹患者の一部には、Y字形の中手骨を伴って多指が第2-4指部に現れる中央列多指症が現れることがあり、そうした例の多くは、その他の症候として口-顔-指症候群Ⅵ型/Varadi-Papp症候群の症候を呈する[Gleesonら2004]。OFDⅥは、現在ではJSの中の1つのタイプとみなされており、molar tooth signに加えて、舌の過誤腫/口腔の小帯/上唇の切痕、中央列多指趾、視床下部過誤腫といった症候の少なくとも1つを有することが要件となる[Porettiら2012]。
端脳梁症候を伴うJoubert症候群(JS-AC)
JSにおいては、脳梁無形成が多くみられる[Valenteら2005]。20人のJS罹患者を対象としたある調査では、ある程度の脳梁異形成が80%に認められている[Senocakら2010]。脳梁の異常は、KIF7の両アレル性病的バリアントを有する例に比較的多くみられ[Bachmann-Gagescuら2015a]、先端脳梁症候群(「遺伝子の上で関連のある疾患」の項を参照)と症候が重なる。先端脳梁症候群では、多指趾や水頭症もみられる[Putouxら2011]。
Jeune呼吸不全性胸郭異形成症候を伴うJoubert症候群(JS-JATD)
JATD(「遺伝子の上で関連のある疾患」の項を参照)、ならびに関連する短肋骨胸郭異形成症であるMainzer-Saldino症候群の症候を有するJS罹患児数人が報告されており、これらの疾患が繊毛起源という共通項を有していることを反映するものとなっている[Lehmanら2010,Halbritterら2013,Shaheenら2015b]。
JSでみられる特定のサブタイプ特異的でないその他の所見
脊柱側彎
生後早期に筋緊張低下がみられる例を中心に、脊柱側彎の報告がみられる。
内分泌異常
内分泌異常の報告がみられる。具体的には、単発性の成長ホルモン分泌不全や甲状腺ホルモン分泌低下から、より広範な汎下垂体機能低下症、あるいは男性における小陰茎といったものが含まれる[Delousら2007,Wolfら2007,Parisiら2009,Sandersら2015]。
肥満
JSでは肥満の増加がみられるようで、同じく繊毛病であるBardet-Biedl症候群との関連が示唆される。JS、MORM症候群(精神発達遅滞mental retardation;肥満obesity;網膜ジストロフィーretinal dystrophy;小陰茎micropenis)の両方でINPP5Eの両アレル性病的バリアントが確認されていることも、こうした関連性を裏打ちするものとなっている[Bielasら2009,Jacobyら2009]。
特徴的顔面症候
側頭骨間距離の狭小化を伴う長い顔、高い眉毛、眼瞼下垂、目立つ鼻梁と上向きの鼻孔、三角形の口、下顎前突、耳介低位といった特徴的顔面症候が時に報告されている[Mariaら1999a](図2A)。ただ、こうした症候は乳児期には認識しづらく、また、今のところ特異的なものとはみなされていない。それでも、現実には「Joubert症候群顔貌」が多く報告されている[Braddockら2007]。KIF7の両アレル性病的バリアントを有する例については、大頭、前額部の突出、眼間開離、高口蓋、小下顎といった頭蓋顔面症候がしばしば現れる[Dafingerら2011,Putouxら2011]。
心疾患
多数のJS罹患者で心疾患が報告されている。一部、口-顔-指症候群の症候に伴う形で現れるような例がみられ、具体的には、中隔欠損、大動脈弁奇形、大動脈縮窄などがある[Bachmann-Gagescuら2015a]。
偏側性の異常
一部の罹患者に、内臓逆位のような偏側性の異常がみられる[Parisiら2009]。
Hirschsprung病
Hirschsprung病が少数例で報告されている[Brancatiら2010]。
難聴
中耳感染症に起因して伝音性難聴がみられることがある[Kroesら2010]。感音性難聴の報告もみられる。
舌肥大
多くの例でリズミックな舌運動がみられ、後に舌肥大に至る場合がある。
他の中枢神経系奇形
- 小脳半球肥大[Porettiら2017]、小脳異所性灰白質[Saraiva & Baraitser 1992]、小脳小葉の配列不整[Senocakら2010,Porettiら2011]。
- Dandy-Walker奇形類似の第四脳室や後頭蓋窩への髄液の異常貯留。これが罹患者の約10%にみられたとする調査[Mariaら2001]、42%近くにみられたとする調査[Porettiら2017]が存在する。
- 後頭部脳瘤(図2E)あるいは髄膜瘤[Genelら2004]。
- 特に口-顔-指症候群Ⅵ型関連所見を示す例における脳幹の異常や視床下部の過誤腫[Porettiら2011]。
- 脳室拡大[Quislingら1999,Senocakら2010,Putouxら2011]、あるいは、Dandy-Walker奇形の古典的徴候を示さず、シャント手術を要する水頭症[Genelら2004]。
- 海馬奇形/回転異常。これは、少数例を対象とした1調査で罹患者の80%、2つのこれより大規模な調査で15%-18%に認められている[Porettiら2011,Porettiら2017]。
- 異所性灰白質、皮質形成異常、脳回肥大、多小脳回[Gleesonら2004,Dixon-Salazarら2004]、神経上皮性嚢胞[Marshら2004,Senocakら2010]などの皮質奇形。
- 神経病理学的評価で確認される橋、小脳、延髄の核・伝導路の異常[Dohertyら2009]、拡散テンソル画像で確認される皮質脊髄路や上小脳路の交叉の欠如[Porettiら2007]、機能的MRIで確認される運動タスク時の活性化パターンの異常[Parisiら2004b]。
遺伝子ごとにみた表現型との相関
遺伝型-表現型相関を述べる上での予備的情報を表3に示した。
表3:表現型に現れる症候ごとにみたJS関連遺伝子
| 遺伝子 | Molar tooth sign以外の表現型上の症候 | 同一アレル疾患/関連疾患3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 網膜ジストロフィー | コロボーマ1 | 腎 | 眼・腎2 | 肝1 | 口 | 多指趾 | その他 | ||
| AHIS1 | ++4 | (+) | +5 | + | (+) | 多小脳回6 | |||
| CPLANE1 | (+) | +7 | +7 | フランス系カナダ人8ならびにオランダ人集団9における創始者効果 | 口-顔-指症候群Ⅵ型 | ||||
| CC2D2A | + | + | + | + | +10 | 脳瘤,脳室拡大,癲癇発作11;フランス系カナダ人集団では軽度の表現型12 | Meckel症候群13 | ||
| CEP290 | ++ | + | ++ | ++14 | + | 脳瘤;心疾患;内臓逆位;その他15 | Leber先天黒内障,Meckel症候群,Bardet-Biedl症候群 | ||
| CSPP1 | (+) | (+) | (+) | (+) | 感音性難聴;脳梁低形成;脳瘤;フッター派集団における創始者効果16 | Meckel症候群,Jeune呼吸不全性胸郭異形成症17 | |||
| INPP5E | + | + | + | (+) | + | MORM症候群18 | |||
| KIAA0586 | (+) | + | (+) | (+) | 幅広い表現型:重度のhydrolethalus症候群(と口蓋裂)から、短肋骨と狭い胸郭を伴うJeune呼吸不全性胸郭異形成症、そして純粋型JSまで19 | hydrolethalus症候群,Jeune呼吸不全性胸郭異形成症 | |||
| MKS1 | (+) | 腎/肝所見と多指趾が1例で報告されている20 | Meckel症候群 | ||||||
| NPHP1 | + | ++ | + | 「軽度のmolar tooth」が時に報告されている21 | 若年性ネフロン癆1型,Cogan症候群 | ||||
| RPGRIP1L | (+) | (+) | ++ | + | (+) | (+) | 脳瘤 | Meckel症候群,網膜疾患22 | |
| TCTN2 21 | 内反足23 | Meckel症候群24 | |||||||
| TMEM67 | +25 | + | ++26,27 | (+) | 脳瘤 | Meckel症候群28 | |||
| TMEM216 29 | (+) | (+) | ++ | + | (+) | + | + | 心所見;脳瘤 | Meckel症候群 |
(+)=多くはないものの報告されている症候;+=一部の例に現れる症候;++=主要症候;MORM=精神発達遅滞mental retardation,体幹型肥満truncal obesity,網膜ジストロフィーretinal dystrophy,小陰茎micropenis
- COACH症候群(小脳虫部低形成cerebellar vermis hypoplasia,精神発達遅滞oligophrenia,運動失調ataxia,コロボーマcoloboma,肝線維症hepatic fibrosis)が含まれる場合あり。
- 「眼・腎」とは、網膜疾患に加えて腎疾患がみられることを指す。過去に用いられた用語に、Senior-Løken症候群(網膜症と若年発症性ネフロン癆)とDekaban-Arima症候群(網膜症,嚢胞性異形成腎)がある。
- 詳しくは「遺伝子の上で関連のある疾患」の項を参照されたい。
- AHI1の両アレル性病的バリアントを有する例に最も多くみられるのが網膜ジストロフィーで、約80%にみられる[Valenteら2008]。早期発症型の先天性盲[Valenteら2006a]、肝疾患[Vilbouxら2017]の報告もみられる。
- ネフロン癆に一致した腎疾患も報告されている[Parisiら,Utschら2006]。
- Dixon-Salazarら[2004],Gleesonら[2004]
- 表現型は、純粋型・古典型Joubert症候群に最も近似しており、一部の罹患者に軸前性・軸後性・中央列の多指趾がみられ、網膜病変[Srourら2015]、肝病変[Vilbouxら2017]を有する例も数例報告されている。罹患者(1.5歳-52歳)に腎障害や肝疾患を示すデータはみられなかったとするものもある[Srourら2012a,Srourら2012b,Srourら2015]。この遺伝子の病的バリアントも口-顔-指症候群Ⅵ型を引き起こし、その典型症候は、軸前性ないし中央列の多指趾、視床下部過誤腫である[Lopezら2014,Romaniら2015]。
- Joubertら[1969]が最初に報告した家系は、この遺伝子の病的バリアントに起因するJoubert症候群である。ケベック州St.Lawrence地域のフランス系カナダ人内で、数種類の反復性病的バリアントがみられる[Srourら2012b,Srourら2015]。
- Kroesら[2016]
- 肝病変が報告されている[Gordenら2008,Noorら2008]。
- CC2D2Aの病的バリアントを有する例は、脳室拡大と癲癇発作を示すことが多い[Bachmann-Gagescuら2012]。
- Srourら[2012a]
- ヌルアレルはMeckel症候群の表現型を引き起こし、ミスセンスバリアントないしhypomorphic型のバリアントはJSを引き起こす[Tallilaら2008,Mougou-Zerelliら2009]。
- 網膜と腎臓の両方に病変を有する例の50%近くが、CEP290に両アレル性の病的バリアントを有している[Valenteら2008]。
- 表現型のスペクトラムは非常に広く、先天性盲、眼のコロボーマ、腎疾患、脳瘤、心臓の中隔欠損、偏側性の異常などがみられる。
- Shaheenら[2014],Vilbouxら[2017]
- この遺伝子の病的バリアントは、時として網膜症と伝音性難聴を伴う古典型JS[Akizuら2014]からJeune骨格異形成症の症候を伴うJS-JATDの表現型[Tuzら2014]、さらには致死性のMeckel症候群様表現型[Shaheenら2014]に至るまで、広い表現型の幅を示す。細い脳梁、後頭部脳瘤、異所性灰白質の報告もみられる[Akizuら2014,Tuzら2014]。
- Jacobyら[2009]
- この遺伝子の病的バリアントは、比較的症候や病変の軽度な「純粋型」JS[Bachmann-Gagescuらら2015b,Roosingら2015]から、Jeune呼吸不全性胸郭異形成症の症候(小さな胸郭,短肋骨,低身長)をもつもの[Albyら2015,Malicdanら2015]、さらには出生前あるいは周産期に死亡に至る水頭症を伴うhydrolethalus症候群の重度の症候を有するもの[Albyら2015]まで、幅広い繊毛病の表現型スペクトラムの幅を示す。罹患者の多くは、フレームシフトによるトランケーションバリアント、スプライス部位バリアント、ナンセンスバリアントといったものをホモ接合あるいは複合ヘテロ接合で有する。表現型がこのように幅広いため、これを病的バリアントの種類ごとに説明することは難しい。
- Meckel症候群罹患者で報告されている他の重度型のバリアントと異なり、MKS1の病的バリアントに起因してJSが生じた例については、少なくとも一方のバリアントは本来の機能を部分的に維持している(例えば、いくらかの機能を保持するミスセンスバリアント)[Romaniら2014,Slaatsら2016]。罹患者の大多数は、網膜ジストロフィーを伴うことのある古典型JSという比較的軽度の表現型を示す。これ以外に、高エコー輝度腎、肝線維症、軸後性多指趾を示した1例(この遺伝子に病的バリアントを有する9人中の1例)が報告されている[Slaatsら2016]。
- NPHP1の両アレル性病的バリアントを有するJS罹患者の一部は、長いながらも細い上小脳脚と比較的軽度の虫部低形成といった特徴的molar tooth signを示す[Parisiら2004a]。(訳注:「TCTN2」にも「21」の番号が振られているが、これは「23」の誤りではないかと思われる。)
- RPGRIP1Lの病的バリアントによってもMeckel症候群が生じる。注目すべきは、より重度の機能喪失型の病的バリアントになるほど、Meckel症候群の表現型が重度になる(多くの場合、致死性になる)ということである[Delousら2007,Wolfら2007]。
- この遺伝子の病的バリアントを有する例の報告は少数にとどまっている。そのため、表現型のスペクトラムについてはよくわかっていない部分がある[Sangら2011]。
- Shaheenら[2011]
- 肝病変の有無とは無関係に、眼のコロボーマを呈する例の53%が、TMEM67の両アレル性病的バリアントを有していた[Dohertyら2010]。
- 肝病変を伴うJS罹患者全体の70%を、TMEM67の両アレル性病的バリアントが占めていた[Dohertyら2010,Iannicelliら2010]。
- Baalaら[2007],Brancatiら[2009],Dohertyら[2010],Vilbouxら[2017]
- 複数の致死型Meckel症候群罹患者に、TMEM67のより重度の機能喪失型バリアントが同定されている[Smithら2006]。一方、部分的に機能が残存するバリアントのほうは、肝疾患を伴うJS、あるいは、molar tooth signその他の神経症状を伴わないネフロン癆と肝線維症の例で同定されている[Ottoら2009,Dohertyら2010]。
- ネフロン癆と多指趾が多くみられる。一部に、口-顔-指症候群の症候を示す例もみられる[Edvardsonら2010,Valenteら2010]。
疾患名について
「Joubert症候群とその関連疾患」(JSRD)という言葉は、過去に、molar tooth signと古典型Joubert症候群の臨床症候を共有し、かつ、その他の器官にも何らかの病変を有する疾患群を指す用語として用いられてきた。紛らわしく、また使用法も一貫していない人名を冠した病名への依存を減らす流れの中で、JSの疾患名については、3つの主要所見を共有する少なくとも8つの臨床的サブタイプに分けるやり方が提唱されてきた(表2)[Brancatiら2010]。そして、「Joubert症候群」という名称は、最近では、JSのすべてのタイプを包括的に指す用語として受容されるようになってきている。
以下に挙げる疾患のいくつかは、過去においては、別個の独立した症候群として報告されていた。しかし、最近の研究で、これらの疾患の罹患者の多くにmolar tooth signがみられることがわかってきた[Satranら1999,Gleesonら2004]。そうした常染色体潜性遺伝疾患の例としては、次のようなものがある。
- Dekaban-Arima症候群(網膜症,嚢胞性異形成腎)[Dekaban 1969]
- Senior-Løken症候群(SLS;網膜症と若年発症性ネフロン癆)[Løkenら1961,Seniorら1961]
- COACH症候群(小脳虫部低形成,精神発達遅滞,運動失調,コロボーマ,肝線維症)[Verloes & Lambotte 1989,Gentileら1996]
・Varadi-Papp症候群(口-顔-指症候群Ⅵ型[OFDⅥ])
これは、小脳虫部低形成、口腔の小帯、舌の過誤腫、正中口唇裂、Y字形の中手骨を伴う特徴的な中央列の多指を示す[Münkeら1990]。腎臓や心臓の病変も報告されている。
発生頻度
Joubert症候群(JS)の発生頻度は明らかになっていない。多くの研究者は80,000人に1人から100,000人に1人の間の数字を挙げているが、この数字は過小評価である可能性がある[Kroesら2007,Parisiら2007,Brancatiら2010]。フランス系カナダ人集団におけるJSの発生頻度は比較的高く、いくつかの創始者バリアントが特定されている。Joubertら[1969]によって最初に報告された家系は、1600年代にフランスからケベックに移住した1人の創始者にまですでに追跡がなされている[Badhwarら2000]。ただ、フランス系カナダ人集団では、この家系をはじめとするいくつかの家系に、CPLANE1の病的バリアントを含む複数のハプロタイプが存在する模様である。実際、フランス系カナダ人の35家系中33家系(94%)において、以下の遺伝子の病的バリアントが同定されている(括弧内は罹患家系の数)。CPLANE1(14),CC2D2A(9),NPHP1(3),TMEM231(2),CEP290(1),TMEM67(1),TCTN1(1),OFD1(1),B9D1(1),C2CD3(1),CEP104(1)。
フランス系カナダ人の多くは、CPLANE1、CC2D2A、TMEM231、NPHP1のいずれかの病的バリアントを複合ヘテロ接合の形で有している[Srourら2012a,Srourら2012b,Srourら2015]。
オランダ人集団においては、これとは異なるCPLANE1の創始者バリアントであるp.Arg2904Terがみられる[Kroesら2016]。
アシュケナージ系ユダヤ人集団においては、TMEM216の創始者バリアントであるp.Arg73Leuの保因者頻度が、92人に1人から100人に1人の割合に上る[Edvardsonら2010,Valenteら2010]。
カナダのフッター派集団においては、脳瘤や嚢胞腎といったMeckel症候群(MKS)類似の表現型を有し、互いに血縁関係にある10人に、TMEM237の病的ナンセンスバリアント(c.52C>T;p.Arg18Ter)のホモ接合が特定されており、この集団における保因者頻度は6%と考えられている[Huangら2011]。また、フッター派Smiedeleut(訳注:フッター派の中の1分派)集団中の別々の2家系で、CSPP1の全く同じ病的フレームシフトバリアント(c.363_364delTA)のホモ接合が確認されている[Shaheenら2014]。これは、上記とは別の創始者バリアントである。
日本人のJS家系の調査では、27家系中6家系にCEP290の病的バリアント(計12の疾患アレル中の9つがc.6012-12T>A)がみられた。27家系中7家系はTMEM67の病的バリアントであったが、こちらについては創始者バリアントを示すアレルは特定されなかった[Suzukiら2016]。
遺伝学的に関連のある疾患(同一アレル疾患)
Joubert症候群(JS)を引き起こす遺伝子の病的バリアントは、同時に、JSと一部の症候が重なる他の疾患でも同定されている。そのため、これまで独立した1疾患と考えられてきたものが本当にJSとは別の存在(すなわち、同一アレル疾患)なのか、それともJSのスペクトラム(表3参照)の一部に属するものなのかということを明確にすることが困難な例が多発している。以下に、そうした疾患群のいくつかを簡単に解説する。
先端脳梁症候群(ACLS)(OMIM 200990)
先端脳梁症候群は常染色体潜性遺伝性疾患で、小頭症、知的障害、脳梁無形成に加え、時に後頭蓋窩の異常、眼間開離、軸前・軸後両方の多指、軸前性多趾を特徴とする。以前より、ACLSとhydrolethalus症候群は同一アレル疾患ではないかと考えられてきた。その後、この両疾患のいくつかの家系においてKIF7の病的バリアントが同定されたことで、そうした推測が確認されるに至った。注目すべきことに、ACLS発端者のうちの数人は、頭蓋の画像診断でmolar tooth sign(MTS)を示す。このことから、ACLSとJSは互いに重なり合う繊毛病であることが示唆されている[Putouxら2011]。
Bardet-Biedl症候群(BBS)
Bardet-Biedl症候群は、通常、常染色体潜性の継承を示し、網膜の錐体杆体ジストロフィー、体幹型肥満、軸後性多指趾、高次脳機能障害、男性の低ゴナドトロピン性性腺機能低下、女性の性器奇形、構造奇形を含めた腎疾患、腎低形成、水腎症、嚢胞腎、糸球体腎炎を特徴とする。進行性の網膜障害により失明に至ることも多く、腎不全により病状が重大になることもある。一部の罹患者には肝線維症がみられる。罹患者の多くは協調運動の障害を伴った運動失調を呈するものの、小脳の病変や構造奇形はふつうみられない[Baskinら2002]。少なくとも19の遺伝子の病的バリアントが報告されており、そのいずれもが一次繊毛に関係するものである。CEP290、MKS1、NPHP1については、BBSとJSの両方の原因遺伝子であることがわかっている[Leitchら2008,Zaghloul & Katsanis 2009,Knoppら2015]。
Cogan症候群(OMIM 257550)
Cogan症候群は、常染色体潜性で家族性の先天性眼球運動失行で、水平方向の随意眼球運動障害を特徴とする。眼球運動失行はJSでも多くみられる症候である。神経線維を追跡する詳細な神経画像において、Cogan症候群とJSとでは走行経路のいくつかに微妙な違いがある可能性が示唆されている[Merliniら2010]。
Cogan症候罹患者の中には、molar tooth signを伴う小脳虫部低形成を示す例がみられ[Whitselら1995,Sargentら1997]、また時に、ネフロン癆を示す例もみられる。NPHP1の290kbの欠失のホモ接合、あるいは290kbの欠失とNPHP1の配列バリアントとの複合ヘテロ接合が、Cogan症候群罹患者の一部で同定されている[Saunierら1997,Betzら2000]。
Hydrolethalus症候群(HLS)(OMIM PS236680)
Hydrolethalus症候群は、致死性の常染色体潜性遺伝疾患で、脳正中部の奇形(鍵穴状大孔を伴う水頭症あるいは無脳症であることが多い)、小下顎、軸後性多指、軸前性多趾などがみられる。フィンランド人集団で、HYLS1の病的バリアントが同定されている[Meeら2005]。血族結婚のアルジェリア人家系で、HLSに一致した症候に加え、molar tooth sign類似の中脳-後脳奇形を有する罹患胎児4人が確認されており、そこではKIF7の病的バリアントが同定されている[Putouxら2011]。また、HLSの胎児では、JSをはじめとするさまざまな繊毛病表現型の罹患者と同様、KIAA0586の病的バリアントも報告されている[Albyら2015]。
Jeune呼吸不全性胸郭異形成症(JATD)
Jeune呼吸不全性胸郭異形成症は、細長い胸郭、低身長、短肢、多指趾、嚢胞性腎疾患に加えて、手足の円錐形骨端、骨幹端の不整、長骨の短小化、三尖形の寛骨臼といった骨格所見を呈する常染色体潜性遺伝の骨系統疾患である。罹患者は、呼吸不全により乳児期に死亡することが多い。これまでに12を超える繊毛関連遺伝子/座位が同定されている(いくつかの繊毛内輸送タンパク質を含む)。複数のTTC21Bのヘテロ接合性病的バリアントがJATDの3家系で同定されており、そのうちの1人の発端者は、ヌルアレルとhypomorphicアレルとの複合ヘテロ接合であった[Davisら2011]。CSPP1[Tuzら2014]とKIAA0586[Malicdanら2015]の病的バリアントについては、JS罹患者とJATDの症候を示す罹患者の両方で同定されている。
Leber先天黒内障(LCA)
Leber先天黒内障は、重度の網膜ジストロフィーで、通常、満1歳までに明らかになる。視機能は一般に不良で、眼振、瞳孔反応の鈍化あるいはほぼ消失、羞明、強度遠視、円錐角膜といったものを伴うことが多い。視力は、20/400(訳注:日本でいう0.05のこと)を上回ることは稀である。特徴的所見として、Franceschettiの眼-指徴候がある。これは、眼を指で突く、押す、擦るといった動作をいう。眼底所見はきわめて多様である。網膜は、最初は異常なしのようにみえることもあるが、しばしばその後、小児期に網膜色素変性症を思わせる色素性の網膜症が出現する。網膜電図は、特徴的な「反応なし」あるいは重度反応低下を示す。LCAの原因として、17以上の遺伝子の病的バリアントが知られている。CEP290の病的バリアントがLCAの約20%を占めるが、ヨーロッパの単発性先天性盲のコホートにおいては、あるイントロンの病的バリアントのホモ接合が少なくとも20%を占めていた[den Hollanderら2006]。
Mainzer-Saldino症候群(MZSDS)
Mainzer-Saldino症候群は、網膜ジストロフィー、腎疾患(通常はネフロン癆)、円錐形の指節骨骨端の3つが診断基準となっている常染色体潜性遺伝疾患である。小脳低形成、狭い胸郭、肝線維症、長頭など、JATDと大きく重なる症候をはじめ、その所見には大きな幅がみられる。IFT140の病的バリアントは、MZSDSとJATDの両方で報告されている[Mainzerら1970,Perraultら2012]。IFT172も同じく繊毛内輸送装置をコードしているが、この遺伝子の病的バリアントがJATD、MZSDS、JSのいずれにおいても報告されている[Halbritterら2013]。
Ellis-van Creveld症候群(EVC)、短肋骨多指症候群(SRPS)、JATD、MZSDSを包括する名称として、多指を伴う、あるいは伴わない短肋骨胸郭異形成(SRTD)(OMIM PS208500)が用いられてきた。ここに属する疾患群は、いずれも常染色体潜性の骨格関連繊毛病で、狭い胸郭、短肋骨、長骨の短縮、「三尖形」の寛骨臼を特徴とする。こうした骨系統疾患とJSの一部の型との間に、大きな症候の重なりがあることは明白である。
Meckel症候群(MIM PS249000)
Meckel症候群は、嚢胞性腎疾患、後頭蓋窩の異常(通常は後頭部の脳瘤)、肝線維症や胆管増生をきたすことになる胆管板奇形を3主徴とする常染色体潜性遺伝疾患である。多指趾も比較的多くみられる。また、一部の罹患者では小脳虫部低形成も報告されている。Meckel症候群は、通常、出生前・周産期致死性である[Kyttäläraら2006,Smithら2006]。Meckel症候群では、これまでに21以上の遺伝子の病的バリアントが同定されている[Knoppら2015]。これらの遺伝子のうちの少なくとも18(CEP290,TMEM67,RPGRIP1L,CC2D2A,CEP41,MKS1,B9D1,B9D2,TMEM138,TMEM231,TCTN2,TCTN3,TMEM237,CPLANE1,CSPP1,CEP120,TMEM107,TMEM216)の病的バリアントについては、JS罹患者でも同じく同定されている[Parisi 2009,Valenteら2010,Thomasら2012,Romaniら2014,Bachmann-Gagescuら2015a,Knoppら2015,Shaheenら2015a,Roosingら2016a,Slaatsら2016]。多くの例で、転写の終止やヌルバリアントといったより重度の影響が予測される病的バリアントでは、致死性のMeckel症候群の表現型が生じ、ミスセンスバリアントのようなより軽度の病的バリアントではJSが生じる[Romaniら2014,Slaatsら2016]。同一家系内の同一の病的バリアントが、Meckel症候群の胎児においてもJSの小児においてもみられるといった例が数家系存在することは、この2つの疾患が1つのスペクトラム内にある可能性を鮮やかに示している[Valenteら2010]。
MORM症候群(OMIM 610156)
MORM(精神発達遅滞mental retardation,体幹型肥満truncal obesity,網膜ジストロフィーretinal dystrophy,小陰茎micropenis)症候群は、Bardet-Biedl症候群と関連があると思われる常染色体潜性遺伝疾患で、INPP5Eの病的バリアントに起因して生じる[Bielasら2009,Jacobyら2009]。この疾患の罹患者は、成長パラメーターの値や寿命は正常で、先天性で非進行性の網膜ジストロフィーと、軽度から中等度で、変化のみられない高次脳機能障害を有する。Bardet-Biedl症候群とは対照的に、多指趾、明らかな性腺機能不全や腎疾患は伴わない[Hampshireら2006]。
ネフロン癆
ネフロン癆は、腎尿細管萎縮と、後に髄質嚢胞の発生に至る進行性の間質線維化を特徴とする常染色体潜性遺伝の腎疾患で、少なくとも19の遺伝子の病的バリアントによって引き起こされる[Hildebrandtら2009,Hurd & Hildebrandt 2011,Wolf 2015]。末期腎疾患の発症年齢には幅がみられ、それにより乳児型、若年型、思春期型といったサブタイプに分かれる。NPHP1の約290kbの欠失のホモ接合が、若年型ネフロン癆罹患者の約25%に認められ[Hoefeleら2005,Saunierら2005,Hildebrandtら2009]、同時に、JS罹患者中の小さなサブセットについては、この欠失が原因となっている。
注:最も多くみられるタイプである若年型ネフロン癆が、JSにおける腎症状の1つとして現れることがある。逆に、ネフロン癆罹患者の10%は、腎以外の症候も併せて有し、それがmolar tooth signであるという場合もある[Saunierら2005]。
口-顔-指症候群(OFD)
口-顔-指症候群は、顔面症候、口腔症候(分葉舌や口腔の小帯であることが多い)、多指趾をはじめとする指趾の奇形を特徴とする、異質性をもった一連の疾患群である。関連してみられる他の臨床症候をもとに、これまでに少なくとも13の臨床的サブタイプが報告されている。そうした各症候は、Meckel症候群、短肋骨多指症候群、JSと大きく重なる部分がある。OFDの原因遺伝子としてこれまでに同定されているものは、すべて繊毛の機能に関連するものであり、JSと重複する遺伝子もいくつか存在する。
口-顔-指症候群Ⅰ型(OFD1)
口-顔-指症候群Ⅰ型は、一次繊毛の機能障害に起因するもので、以下のような異常を特徴とする。
- 口(分葉舌,舌の過誤腫あるいは脂肪腫,硬口蓋あるいは軟口蓋の裂,歯肉部の余剰小帯,無歯症,その他の歯の異常)
- 顔(眼間開離あるいは内眼角開離,鼻翼低形成,上口唇正中の口唇裂あるいは偽口唇裂,小下顎)
- 指(短指趾,種々の程度の合指趾,第5指の彎指,重複拇趾,軸前性あるいは軸後性多指)
- 脳(脳内嚢胞,脳梁無形成,Dandy-Walker奇形を伴うことのある小脳無形成)
- 腎(多発性嚢胞腎)
ふつうは軽度であるが、OFD1罹患者の50%近くにある程度の知的障害がみられる。罹患者は、ほぼすべて女性である。OFD1の男性例も報告されてはいるものの、OFD1の女性が産んだOFD1の男性胎児といった程度の報告にとどまっている。
注目すべきこととして、4例で報告された新たな臨床症候(胎児水腫,黄疸,深部腱反射の反射活発,癲癇発作,三葉化左肺)[Terespolskyら1995,Brzustowiczら1999]がOFD1の病的バリアントに起因するものであることが認識されるようになったことで、OFD1の表現型のスペクトラムが大きく広がったことが挙げられる[Budnyら2006]。OFD1の病的バリアントは、OFDの症候を有する男性JSという稀な例においても報告されている[Coeneら2009,Fieldら2012]。
口-顔-指症候群Ⅳ型(OFDⅣ,Mohr-Majewski症候群)(OMIM 258860)
口-顔-指症候群Ⅳ型は、拇趾と軸後性の多合指趾、脛骨異形成、さまざまな程度の短肋骨、嚢胞腎、脳奇形を特徴とする。重度で致死性のOFDⅣ表現型を示し、長骨の彎曲、嚢胞腎、後頭部脳瘤、肝臓の胆管増生を有するものの短肋骨は呈しないいくつかの血統において、TCTN3のトランケーション型病的バリアントが同定されている。こうした胎児の中には、molar tooth signを示唆するような虫部無形成を呈する例もみられている[Thomasら2012]。
この表現型がMeckel症候群やJSと症候が重なるという点に、注目が必要である。
口-顔-指症候群Ⅵ型(OFDⅥ,Varadi-Papp症候群)(OMIM 277170)
OFDⅥ罹患者には、中央列の指趾の間に過剰の指趾が現れる中央列多指趾とY字形中手骨、小脳虫部低形成、口腔の小帯、分葉舌あるいは舌の過誤腫(図2B)、それに眼間開離や口唇正中部の溝といった頭蓋顔面症候がしばしばみられる。腎臓や心臓の病変も報告されている[Münkeら1990]。そして結果として、咀嚼障害、嚥下障害、呼吸の問題が生じることがある。OFDⅥはJSの中の1つの型と捉えられてきており、molar tooth signに加え、舌の過誤腫/上口唇の切痕、中央列多指趾、視床下部過誤腫の1つ以上を有することが要件とされている[Porettiら2012]。ある研究グループは、OFDⅥを有する11家系中9家系においてCPLANE1の病的バリアントを検出している[Lopezら2014]。ただ、軸前性ないし中央列の多指趾と視床下部過誤腫については、CPLANE1の病的バリアントに起因して生じている模様であったが、舌の過誤腫と舌の小帯については、この遺伝子の病的バリアントに起因するものではなかった[Lopezら2014,Romaniら2015]。一方、別のある研究グループによると、CPLANE1の病的バリアントが検出されたのはOFDⅥ罹患者17人中2人にとどまっていたとした上で、OFDⅥについては、TMEM216、TMEM107、OFD1の病的バリアントもみられたと報告している[Romaniら2015,Lambacherら2016]。
鑑別診断
鑑別診断の対象となる疾患は、すでに「遺伝子の上で関連のある疾患」の項で述べた疾患群である。
臨床的マネジメント
最初の診断に続いて行う評価
Joubert症候群(JS)と診断された例については、罹患者である乳児/小児の有する疾患の範囲を特定することを目的として、以下に示すようなベースラインの評価を行うことが勧告されている[Parisiら2007](全文はこちら)。勧告は、「Joubert症候群とその関連疾患の会」のウェブサイトで、その概略を見ることができる。
- 診断時にすでに実施済でなければ、予後不良あるいは癲癇発作の前触れとなる可能性のある脳奇形、神経細胞移動障害、頭瘤といったものを評価するための高性能MRI検査
- 筋緊張、呼吸パターン(頻呼吸と無呼吸)、眼球運動、発達、小脳機能に重点を置いたベースラインとしての神経学的評価
- ベースラインの評価として、また、特に症候として無呼吸がみられるときに行う睡眠ポリグラフを用いた睡眠の検査
- 言語治療士による口腔運動機能の評価、ないしX線透視嚥下検査
- 年齢相当の評価ツールを用いた発達評価
- コロボーマや網膜の変化を調べるための散瞳検査、ならびに斜視や眼瞼下垂の検査といった小児眼科医による評価 視覚誘発電位、網膜電図、眼球運動検査などの特殊検査も考慮
- 肝線維症、腎嚢胞、ないしネフロン癆の所見(例えば、腎皮髄境界の消失)といったものを調べるための腹部超音波検査
- 血圧、血清尿素窒素(BUN)、血清クレアチニン濃度、全血算(CBC)、さらに、可能なようなら濃縮能を調べるための早朝尿を用いた尿比重試験などの尿検査も含めた腎機能検査
- トランスアミナーゼ、アルブミン、ビリルビンの血清濃度、プロトロンビン時間などの肝機能検査
- 小陰茎の男性、あるいは成長ホルモン分泌不全の徴候を有する子どもについて、他の下垂体異常を調べるための内分泌評価
- 短肋骨多指趾やJATDなどの骨格の異常が疑われるときは、骨格の診査ないし四肢のX線写真検査
- 家族歴の聴取、成長や頭囲の評価、ならびに多指趾・顔貌の異常・舌の腫瘍/分葉舌・小陰茎といったその他の奇形を調べるための臨床遺伝医との面談
症状に対する治療
呼吸器
- 呼吸パターンの異常を有する乳幼児・小児については、異常の程度が重度であるなら、無呼吸のモニタリングを検討すべきである。新生児期を中心に、カフェインをはじめとする刺激剤、酸素補給などの支持療法が考えられる。
- 明らかな呼吸障害がみられる乳児に対して行う外科処置の際の麻酔管理については、一部の例では次のようなものを使用することで対応しうる可能がある。
- 無呼吸の悪化防止のため、オピオイドを避けた形で行う局所麻酔[Vodopich & Gordon 2004]。
- オピオイドによる呼吸抑制その他の合併症を回避しつつ静止画像を得ることを目的とした、クロニジンやデクスメデトミジンなどのα2アゴニストの使用[Sriganeshら2004]。
- 稀ながら、重度の呼吸障害を有する子どもについては、人工呼吸や気管切開が検討されることもある。
- 中耳感染症に対しては、伝音性難聴を回避するため、積極的治療が行われる。
筋緊張低下とそれに対する治療的介入
- 口腔運動機能異常に対する言語治療士による適切な管理と治療
- 重度の嚥下障害を有する子どもについては、経鼻胃管あるいは胃瘻管での栄養
- 早期介入プログラムを通じて行う作業療法、理学療法、言語治療
- 学童に対し、学業成績を最大限に伸ばすことを目的とした個別的教育評価や支援
- 適切な年齢から定期的に行う神経心理学的検査や発達検査
他の中枢神経系奇形
- 水頭症の徴候(頭囲の急激な拡大ないし大泉門の膨隆)がみられる場合は、脳神経外科に紹介を行う。
- 注:JSで水頭症が生じた場合、シャント術が必要になることは稀である。
- 後頭蓋窩の嚢胞や髄液貯留に関して介入が必要になることは稀である。
- 脳瘤については、一期的手術での閉鎖が必要となろう。
- 癲癇発作については、標準的な抗痙攣薬を用いて、神経内科医の手で評価や治療を行う。
- Joubert症候群でみられる行動上の合併症の治療には、向精神薬が用いられる。ただし、すべての子どもに一様に有効な薬剤があるわけではない。
眼
- 症状を伴う眼瞼下垂、斜視、弱視については、必要に応じ手術を行う。
- 屈折異常に対しては、屈折矯正用レンズを使用する。
- 眼球運動失行に対しては、ビジョンセラピーも考えられる。ただし、本疾患にこれを応用した研究はあまりみられない。
- 先天性盲あるいは進行性網膜ジストロフィーがみられる場合には、視覚障害者向けの介入を行う。
腎疾患
- 腎臓専門医の診察。
- ネフロン癆に起因する末期腎疾患(ESRD)に対しては、ティーンの時期以降に透析や腎移植が必要となる。
- 高血圧、貧血をはじめとするESRDの合併症については、状況に応じた治療を行う。
肝線維症
- 消化器医の診察
- 肝不全、肝線維症に対しては、食道静脈瘤や門脈圧亢進に対する門脈シャント術を適宜織り交ぜながら、消化器医が管理を行う。
- 一部、同所肝移植が必要になる例がみられる。
骨格
- 多指趾に対する外科的治療
- 脊柱側彎に対する整形外科医による適切な医療的管理
その他
- 口腔顔面裂については、標準的な外科的介入が行われる。
- 舌の腫瘍によって嚥下障害や呼吸障害がみられる場合には、外科的切除が必要になることがある。
- 比較的年長で、睡眠時無呼吸や舌肥大の症候がみられる例については、睡眠ポリグラフによる評価、ないし、アデノイド切除術、口蓋扁桃摘出術、舌縮小術を念頭に置いた耳鼻科医による評価が必要になることがある。一部の小児については、夜間にBiPAPやC-PAPが使用される。
- 月経不順や下垂体ホルモン分泌不全については、(必要に応じ、ホルモン補充療法を念頭に)内分泌内科医の診察が望ましい。
- 肥満については、食餌療法、運動療法、行動療法などで適切に対応する。
- 先天性心疾患や内臓逆位に関しては通常の治療を行う。
・Hirschsprung病がみられる場合は、外科的改善が必要になる。
二次的合併症の予防
構造的心奇形を有する罹患者については、外科的処置や歯科的処置の際に抗生剤の予防投与が必要となる。
定期的追跡評価
Joubert症候群の乳幼児に発生しうる合併症を予測する上で役立つ、広く通用する特定の特徴というものが存在するわけではないので、数多くの点について年に1度評価していくことが推奨される(「Joubert症候群とその関連疾患の会」のウェブサイトも参照)。
- 成長、性的成熟、呼吸(無呼吸の症候を含む)、運動機能に関する小児科的、神経学的評価、ならびにモニタリング
- 必要に応じ、神経心理学的、発達的な評価や検査
- 視力、追視能力、網膜ジストロフィーの発症等に関する眼科的評価
- 発生しうる肝・腎の異常を評価するための腹部超音波検査
- 肝機能検査
- 腎機能の評価:血圧、血清BUN・クレアチニン濃度の測定、CBC、早朝尿を用いた尿検査
避けるべき薬剤/環境
腎障害を有する罹患者については、非ステロイド性抗炎症薬など、腎毒性を有する薬剤を避ける必要がある。
肝障害を有する罹患者については、肝毒性を有する薬剤を避ける。
リスクを有する血縁者の評価
JS罹患者と似た臨床症候を有する同胞や血族については、遺伝科の診察が必要となる。発端者の有する病的バリアントがすでに同定されている場合は、症候を有する血族がこのバリアントを有しているかどうかを検査することが望ましい。
リスクを有する血族に対して行う遺伝カウンセリングを目的とした検査関連の事項については、「遺伝カウンセリング」の項を参照されたい。
研究段階の治療
さまざまな疾患・状況に対して進行中の臨床試験に関する情報については、アメリカの「Clinical Trials.gov」、ならびにヨーロッパの「EU Clinical Trials Register」を参照されたい。
注:現時点で本疾患に関する臨床試験が行われているとは限らない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
Joubert症候群(JS)は、主として常染色体潜性の遺伝形式をとる。
OFD1関連JSについては、X連鎖性の遺伝形式をとる(X連鎖性遺伝の場合の説明についてはリンク先を参照)。(訳注:「血縁者の有するリスク(常染色体潜性遺伝)」の項のすぐ後ろに訳出している)
血縁者の有するリスク(常染色体潜性遺伝)
発端者の両親
- 罹患児の両親は絶対ヘテロ接合者(すなわち、JS関連遺伝子の病的バリアントを1つ有する保因者)である。
- ヘテロ接合者は無症状で、本疾患を発症するリスクは有しない。
発端者の同胞
- 発端者の同胞は、受胎の段階で、罹患者である可能性が25%、無症状の保因者である可能性が50%、罹患者でも保因者でもない可能性が25%である。
- ヘテロ接合者(保因者)は無症状で、本疾患を発症するリスクは有しない。
発端者の子
- 発端者の子は、JS関連遺伝子の病的バリアントに関して絶対ヘテロ接合者となる。
- これまでにJS罹患者が子をもうけたという報告はみられないが、本疾患の高次脳機能障害は幅広いスペクトラムを示すことがわかっており、今後ほどなく、罹患者が子をもうけたという報告がなされる可能性は高まっている。
他の家族構成員
発端者の両親の子は、それぞれ、JS関連遺伝子の病的バリアントの保因者であることに関し、50%のリスクを有する。
X連鎖性遺伝の場合の血縁者の有するリスクについての情報はこちらをクリック(本稿末尾)
血縁者の有するリスク(X連鎖性遺伝 ― OFD1関連)
男性発端者の親
- 男性罹患者の父親は、罹患者でも、OFD1の病的バリアントのヘミ接合者でもない。したがって、父親に関しては、それ以上の評価や検査は不要である。
- 家系内に複数の罹患者がいる場合は、罹患男性の母親は絶対保因者である。
注:1人の女性に複数の罹患児があり、血族内に他の罹患者がいない、そしてさらに、その女性の白血球DNAから病的バリアントが検出されないといった場合、その女性は生殖細胞系列モザイクである可能性が高い。
- 男性が家系内で唯一の罹患者(すなわち、孤発例)である場合は、母親がヘテロ接合者(保因者)であるか、もしくは、罹患男性に生じたOFD1のde novoの病的バリアントということになる。後者の場合、母親はヘテロ接合者(保因者)ではないことになる。
男性発端者の同胞
- 同胞の有するリスクは、母親の遺伝的状態によって変わってくる。
- 発端者の母親が病的バリアントを有していた場合は、妊娠に際してそれを伝達する可能性は50%である。病的バリアントを継承した男児は罹患者となる。一方、病的バリアントを継承した女性が罹患者となる可能性は低い。
- 発端者が孤発例(すなわち、家系内で唯一の罹患者)で、なおかつ、母親の白血球DNAから病的バリアントが検出されない場合、同胞の有するリスクは一般集団よりわずかに高い(それでも1%未満)程度となる。それは、理論上、母親の生殖細胞系列モザイクの可能性が残るからである。
男性発端者の子
- JS罹患者が子をもうけたとする報告はみられない。
- X連鎖性JSの男性は、娘に対しては必ず病的バリアントを伝達することになるが、息子に対してこれを伝達することはない。
他の血縁者
発端者の母方の伯母(叔母)はヘテロ接合者(保因者)であることに関し、リスクを有する。伯母(叔母)の子は、その性別により、ヘテロ接合者(保因者)もしくは罹患者となるリスクを有する。
注:分子遺伝学的検査を行うことで、家系内でde novoの病的バリアントが生じた人を特定できることがある。これが判明することで、家系内のより広い範囲について、遺伝的リスクの確定が可能となる。
保因者の特定
リスクを有する血縁者に対して保因者の検査を行うためには、家系内に存在するJS関連病的バリアントを事前に特定しておく必要がある。
関連する遺伝カウンセリング上の諸事項
早期診断・早期治療を目的としてリスクを有する血縁者に対して行う評価関連の情報については、「臨床的マネジメント」の「リスクを有する血縁者の評価」の項を参照されたい。
家族計画
- 遺伝的リスクの確定、保因者であるかどうかの明確化、出生前/着床前遺伝学的検査を受けるかどうかの話し合いに最も適しているのは、妊娠前の時期である。
- 罹患者、保因者、あるいは、保因者であるリスクを有する若い成人に対しては、遺伝カウンセリング(子に生じる可能性のあるリスクや、子をもうける上での選択肢についての説明を含む)を提供することが望ましい。
DNAバンキング
検査の手法であるとか、遺伝子・病原のメカニズム・疾患等に対するわれわれの理解が、将来はより進歩していくことが予想される。そのため、分子診断の確定していない(すなわち、原因となった病原のメカニズムが未解明の)発端者のDNAについては、保存することを検討すべきである。
出生前診断ならびに着床前遺伝学的検査
分子遺伝学的検査
家系内に存在するJS関連の病的バリアントが同定済の場合は、高リスクの妊娠に備えた出生前診断や、JSに関する着床前遺伝学的検査を行うことが可能である。
出生前診断の利用に関しては、医療者間でも、また家族内でも、さまざまな見方がある。現在、多くの医療機関では、出生前診断を個人の決断に委ねられるべきものと考えているようであるが、こうした問題に関しては、もう少し議論を深める必要があろう。
胎児超音波検査
JSに関して25%の罹患リスクを有する妊娠について、超音波検査を用いて第1三半期に脳瘤をはじめとする脳の構造異常を確認することで診断を行った報告がみられる[van Zalen-Sprockら1996,Wangら1999]。リスクを有する胎児の出生前診断でより一般的なのは、第2三半期に出生前超音波検査で、後頭蓋窩、腎(嚢胞・腎肥大・高エコー腎)、指趾(多指趾)を調べることで診断がなされるといったケースである[Ní Scanaillら1999,Aslanら2002,Dohertyら2005]。実際問題として、JSの胎児にみられる出生前超音波所見は、nuchal transparency、大槽の拡大、小脳虫部無形成/低形成、後頭部の脳瘤、脳室拡大といった比較的非特異的なものであるため、家族歴を有しない場合のJSの確定的な診断は困難である。さらに、小脳虫部は発生が比較的遅く、妊娠18週になって初めて第4脳室を覆うようになるため、妊娠の初期段階でmolar tooth sign(MTS)を視認することは困難である[Bromleyら1994]。2D超音波と、サーフェスレンダリングを用いた3D超音波再構成を併用することで、妊娠22週の段階で、JSの家族歴のない胎児数例のMTSを可視化できたとする報告がみられる[Quarelloら2014]。
JSのリスクを有する胎児に対する正確な出生前診断が、妊娠11週ないし12週から始めて超音波画像診断を連続的に行って、妊娠20週までの小脳その他の胎児の解剖学的構造を詳細に評価するとともに、その後、妊娠20週から22週の間に胎児MRI画像診断を行うことで達成されたとする報告がみられる[Dohertyら2005]。また、胎児がJSであることに関し25%のリスクを有する12の妊娠において、橋中脳境界(MTSを含む領域)の胎児MRI所見をもとに、妊娠22週の段階で3胎児の診断を正しく行えたとする報告が、1医療機関から行われている[Saleem & Zaki 2010]。リスクを有する妊娠において、現在までに報告されている最も早い診断は、妊娠17週から18週に胎児MRIでMTSの同定がなされたとする2例である[Saleemら2011]。胎児MRIを含む出生前画像診断は、後頭蓋窩の奇形の診断には確かに有用ではあるが、JSの診断に関する感度や特異度についてはよくわかっておらず、これを使用することに関する系統立った評価は未だなされていない。
すでにJSの子どもをもっているカップルにとって、Joubert症候群とその関連疾患の出生前診断をうかがわせる所見(例えば、胎児画像診断で確認される脳瘤、腎の嚢胞性変化、多指趾、後頭蓋窩の奇形)の存在は非常に大きな意味をもつものではあるが、画像診断の感度についてはよくわかっておらず、また、家系内のばらつきの幅という要素もあることから、こうした徴候がみられないということが、必ずしもJoubert症候群とその関連疾患の可能性を排除するものにはならない。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- Joubert Syndrome and Related Disorders Foundation
Phone: 614-864-1362
Email: info@jsrdf.org
www.jsrdf.org
- National Institute of Neurological Disorders and Stroke (NINDS)
PO Box 5801
Bethesda MD 20824
Phone: 800-352-9424 (toll-free); 301-496-5751; 301-468-5981 (TTY)
Joubert Syndrome Information Page
- National Library of Medicine Genetics Home Reference
- Apraxia Kids
Phone: 412-785-7072
Email: info@apraxia-kids.org
www.apraxia-kids.org
- Ciliopathy Alliance
United Kingdom
www.ciliopathyalliance.org
- Joubert Syndrome Link to Information & Family Exchange (JS-LIFE Registry)
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A:Joubert症候群:遺伝子とデータベース
| 遺伝子 | 染色体上の座位 | タンパク質 | Locus-Specificデータベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| AHI1 | 6q23.3 | Jouberin | AHI1@LOVD | AHI1 | AHI1 |
| ARL13B | 3q11.1-q11.2 | ADP-ribosylation factor-like protein 13B | ARL13B database | ARL13B | ARL13B |
| B9D1 | 17p11.2 | B9 domain-containing protein 1 | B9D1@LOVD | B9D1 | B9D1 |
| B9D2 | 19q13.2 | B9 domain-containing protein 2 | B9D2 | B9D2 | |
| C2CD3 | 11q13.4 | C2 domain-containing protein 3 | C2CD3 | C2CD3 | |
| CC2D2A | 4p15.32 | Coiled-coil and C2 domain-containing protein 2A | CC2D2A | CC2D2A | |
| CEP41 | 7q32.2 | Centrosomal protein of 41kDa | CEP41 | CEP41 | |
| CEP104 | 1q36.32 | Centrosomal protein of 104kDa | CEP104 | CEP104 | |
| CEP120 | 5q23.2 | Centrosomal protein of 120kDa | CEP120 | CEP120 | |
| CEP290 | 12q21.32 | Centrosomal protein of 290kDa | CEP290 | CEP290 | |
| CPLANE1 | 5p13.2 | Ciliogenesis and planar poliarity effector 1 | C5orf42@LOVD | CPLANE1 | CPLANE1 |
| CSPP1 | 8q13.1-q13.2 | Centrosome and spindle pole-associated protein 1 | CSPP1 | CSPP1 | |
| IFT172 | 2p23.3 | Intraflagellar transport protein 172 homolog | IFT172 | IFT172 | |
| INPP5E | 9q34.3 | Phosphatidylinositol polyphosphate 5-phosphatase Type Ⅳ | INPP5E@LOVD | INPP5E | INPP5E |
| KATNIP | 16p12.1 | Katanin-interacting protein | KATNIP | KATNIP | |
| KIAA0586 | 14q23.1 | Protein TALPID3 | KIAA0586 | KIAA0586 | |
| KIF7 | 15q26.1 | Kinesin-like protein KIF7 | KIF7@LOVD | KIF7 | KIF7 |
| MKS1 | 17q22 | Meckel syndrome type 1 protein | MKS1@LOVD | MKS1 | MKS1 |
| NPHP1 | 2q13 | Nephrocystin-1 | NPHP1@LOVD | NPHP1 | NPHP1 |
| OFD1 | Xp22.2 | Oral-facial-digital syndrome 1 protein | OFD1@LOVD | OFD1 | OFD1 |
| PDE6D | 2q37.1 | Retinal rod rhodopdin-sensitive cGMP 3’,5’-cyclic phosphodiesterase subunit delta | PDE6D | PDE6D | |
| POC1B | 12q21.33 | POC1 centriolar protein homolog B | POC1B | POC1B | |
| RPGRIP1L | 16q12.2 | Protein fantom | RPGRIP1L | RPGRIP1L | |
| TCTN1 | 12q24.11 | Tectonic-1 | TCTN1@LOVD | TCTN1 | TCTN1 |
| TCTN2 | Tectonic-2 | TCTN2 | TCTN2 | ||
| TCTN3 | 10q24.1 | Tectonic-3 | TCTN3 | TCTN3 | |
| TMEM67 | 8q22.1 | Meckelin | TMEM67@LOVD | TMEM67 | TMEM67 |
| TMEM107 | Transmembrane protein 107 | TMEM107 | TMEM107 | ||
| TMEM138 | 11q12.2 | Transmembrane protein 138 | TMEM138 | TMEM138 | |
| TMEM216 | 11q12.2 | Transmembrane protein 216 | TMEM216 database | TMEM216 | TMEM216 |
| TMEM231 | 16q23.1 | Transmembrane protein 231 | TMEM231 | TMEM231 | |
| TMEM237 | 2q33.1 | Transmembrane protein 237 | TMEM237@LOVD | TMEM237 | TMEM237 |
| TTC21B | 2p24.3 | Tetratricopeptide repeat protein 21B | TTC21B | TTC21B | |
| ZNF423 | 16q12.1 | Zinc finger protein 423 | ZNF423 | ZNF423 |
データは、以下の標準資料から作成したものである。
遺伝子についてはHGNCから、染色体上の座位についてはOMIMから、タンパク質についてはUniProtから。
リンクが張られているデータベース(Locus-Specific,HGMD,ClinVar)の説明についてはこちらをクリック。
表B:Joubert症候群関連のOMIMエントリー閲覧はすべてOMIMへ)
| 213300 | JOUBERT SYNDROME 1; JBTS1 |
| 243910 | ARIMA SYNDROME |
| 300170 | OFD1 CENTRIOLE AND CENTRIOLAR SATELLITE PROTEIN; OFD1 |
| 300804 | JOUBERT SYNDROME 10; JBTS10 |
| 602676 | PHOSPHODIESTERASE 6D; PDE6D |
| 604557 | ZINC FINGER PROTEIN 423; ZNF423 |
| 607100 | NEPHROCYSTIN 1; NPHP1 |
| 607386 | INTRAFLAGELLAR TRANSPORT 172; IFT172 |
| 608091 | JOUBERT SYNDROME 2; JBTS2 |
| 608629 | JOUBERT SYNDROME 3; JBTS3 |
| 608894 | ABELSON HELPER INTEGRATION SITE 1; AHI1 |
| 608922 | ADP-RIBOSYLATION FACTOR-LIKE GTPase 13B; ARL13B |
| 609583 | JOUBERT SYNDROME 4; JBTS4 |
| 609863 | TECTONIC FAMILY, MEMBER 1; TCTN1 |
| 609883 | MKS1 TRANSITION ZONE COMPLEX SUBUNIT 1; MKS1 |
| 609884 | TRANSMEMBRANE PROTEIN 67; TMEM67 |
| 610142 | CENTROSOMAL PROTEIN, 290-KD; CEP290 |
| 610178 | KIAA0586 GENE; KIAA0586 |
| 610188 | JOUBERT SYNDROME 5; JBTS5 |
| 610523 | CENTROSOMAL PROTEIN, 41-KD; CEP41 |
| 610688 | JOUBERT SYNDROME 6; JBTS6 |
| 610937 | RPGRIP1-LIKE; RPGRIP1L |
| 611254 | KINESIN FAMILY MEMBER 7; KIF7 |
| 611560 | JOUBERT SYNDROME 7; JBTS7 |
| 611654 | CENTROSOME SPINDLE POLE-ASSOCIATED PROTEIN 1; CSPP1 |
| 611951 | B9 DOMAIN-CONTAINING PROTEIN 2; B9D2 |
| 612013 | COILED-COIL AND C2 DOMAINS-CONTAINING PROTEIN 2A; CC2D2A |
| 612014 | TETRATRICOPEPTIDE REPEAT DOMAIN-CONTAINING PROTEIN 21B; TTC21B |
| 612285 | JOUBERT SYNDROME 9; JBTS9 |
| 612291 | JOUBERT SYNDROME 8; JBTS8 |
| 613037 | INOSITOL POLYPHOSPHATE-5-PHOSPHATASE, 72-KD; INPP5E |
| 613277 | TRANSMEMBRANE PROTEIN 216; TMEM216 |
| 613446 | CENTROSOMAL PROTEIN, 120-KD; CEP120 |
| 613820 | NEPHRONOPHTHISIS 12; NPHP12 |
| 613846 | TECTONIC FAMILY, MEMBER 2; TCTN2 |
| 613847 | TECTONIC FAMILY, MEMBER 3; TCTN3 |
| 614144 | B9 DOMAIN-CONTAINING PROTEIN 1; B9D1 |
| 614173 | JOUBERT SYNDROME 13; JBTS13 |
| 614423 | TRANSMEMBRANE PROTEIN 237; TMEM237 |
| 614424 | JOUBERT SYNDROME 14; JBTS14 |
| 614459 | TRANSMEMBRANE PROTEIN 138; TMEM138 |
| 614464 | JOUBERT SYNDROME 15; JBTS15 |
| 614465 | JOUBERT SYNDROME 16; JBTS16 |
| 614571 | CILIOGENESIS AND PLANAR POLARITY EFFECTOR 1; CPLANE1 |
| 614615 | JOUBERT SYNDROME 17; JBTS17 |
| 614784 | POC1 CENTRIOLAR PROTEIN B; POC1B |
| 614815 | JOUBERT SYNDROME 18; JBTS18 |
| 614844 | NEPHRONOPHTHISIS 14; NPHP14 |
| 614949 | TRANSMEMBRANE PROTEIN 231; TMEM231 |
| 614970 | JOUBERT SYNDROME 20; JBTS20 |
| 615636 | JOUBERT SYNDROME 21; JBTS21 |
| 615665 | JOUBERT SYNDROME 22; JBTS22 |
| 615944 | C2 CALCIUM-DEPENDENT DOMAIN-CONTAINING PROTEIN 3; C2CD3 |
| 616183 | TRANSMEMBRANE PROTEIN 107; TMEM107 |
| 616490 | JOUBERT SYNDROME 23; JBTS23 |
| 616650 | KATANIN-INTERACTING PROTEIN; KATNIP |
| 616654 | JOUBERT SYNDROME 24; JBTS24 |
| 616690 | CENTROSOMAL PROTEIN, 104-KD; CEP104 |
| 616781 | JOUBERT SYNDROME 25; JBTS25 |
| 616784 | JOUBERT SYNDROME 26; JBTS26 |
| 617120 | JOUBERT SYNDROME 27; JBTS27 |
| 617121 | JOUBERT SYNDROME 28; JBTS28 |
| 617761 | JOUBERT SYNDROME 31; JBTS31 |
分子レベルの病原性
Joubert症候群(JS)を引き起こすことが知られている遺伝子は、すべて、繊毛の形成、形態、機能に一定の役割を果たすものと考えられる一次繊毛ないし基底小体(basal body)・中心体(centrosome)に関連するものである。繊毛は、膜から立ち上がった毛髪様突起で、その基部は基底小体に固定されている。
運動性繊毛は、中心部の2本、プラス2本組の微小管9組でできた軸糸構造(9+2)を有し、これにより運動性と液体の流動性が確保されており、呼吸上皮や精子などの特定の細胞種でみられる。これに対し、一次繊毛は9+0の微小管構造で、通常は非運動性である。一次繊毛はほとんどの細胞腫にみられ、細胞の化学的・機械的感覚受容や、細胞分化・細胞分裂・平面内細胞極性といったものに関係するWNT、ソニックヘッジホッグ(SHH)、PDGFなどの細胞のシグナル伝達に一定の役割を果たしているように見受けられる。
繊毛病は、繊毛機能に重要な数多くのタンパク質の中のいくつかに生じた障害に起因する疾患群で、それらに共通する症候としては、腎疾患、網膜ジストロフィー、多指趾などがある[Badanoら2006による総説がある]。繊毛の異常と表現型との関係については、未だ十分には解明されていないものの、Joubert症候群でみられる後脳奇形を有する例について言うと、神経管の背腹軸パターン形成と小脳顆粒細胞の増殖の両方にSHHシグナル伝達が決定的に重要であることが知られている[Doherty 2009]。
注:ここからのセクションでは、病的バリアントがJS全体の中で1%を占めるJS関連遺伝子(表1a参照)について詳細を述べる。JS全体の中に占める割合が1%未満のJS関連遺伝子(表1b参照)については、リンク先で別に詳述する(訳注:本編の後ろに訳出している)。
AHI1
遺伝子構造
AHI1は28のエクソンから成り、選択的スプライシングによるいくつかのバリアントが存在する。最も一般的な全長転写物は5,528bpである。
病的バリアント
これまでに、ナンセンス・ミスセンス・スプライス部位バリアント、欠失、挿入のホモ接合が報告されている[Dixon-Salazarら2004,Ferlandら2004,Parisiら2006,Romanoら2006,Utschら2006](より詳細な情報については、表Aの「Locus Specificデータベース」の項を参照)。
正常遺伝子産物
正常遺伝子産物は、1,196のアミノ酸から成るAHI1(jouberinとも呼ばれる)である。このタンパク質は、1つのコイルドコイルドメイン、1つのSH3ドメイン、6つのWD40リピートを有し、シグナル伝達、RNAプロセシング、小胞輸送などのさまざまな機能を担っているものと推定されている。
異常遺伝子産物
AHI1の機能喪失は、Joubert症候群を引き起こす。生存Ahi1ヌルマウス株における表現型は、周産期死亡型から、視細胞感覚繊毛と視細胞外節の正常発生の障害を伴う網膜変性の早発に至るまで、幅がみられる[Westfallら2010]。
CPLANE1
遺伝子構造
CPLANE1の参照配列(NM_023073.3)は、53のエクソンから成り、3,197のアミノ酸から成るタンパク質(NP_075561.3)をコードする[Srourら2012b,Srourら2015]。
病的バリアント
互いに血縁関係のない数多くのフランス系カナダ人家系の14人から、8種類の病的バリアントが確認されている。そのうちのいくつかについては、フランス系カナダ人集団内における創始者効果を示す1つの特定のハプロタイプと連鎖している[Srourら2012b,Srourら2015]。多くの罹患者は、2種類の病的バリアントを複合ヘテロ接合で有している。これとは別に、オランダ人集団でも創始者バリアント(p.Arg2904Ter)が報告されている[Kroesら2016]。この遺伝子の病的バリアントは、OFDⅥの表現型を示す例においても報告されている[Lopezら2014,Romaniら2015]。
表4:このGeneReviewで取り上げたCPLANE1の病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.4006C>T | p.Arg1336Trp | |
| c.4804C>T | p.Arg1602Ter | NM_023073.3 |
| c.6354dupT | p.Ile2119TyrfsTer2 | NP_075561.3 |
| c.6407delC | p.Pro2136HisfsTer31 | |
| c.7400+1G>A | NM_02373.3 | |
| c.7477C>T | p.Arg2493Ter | NM_023073.3 |
| c.8710C>T | p.Arg2904Ter | NP_075561.3 |
| c.4690G>A | p.Ala1564Thr | 脚注1参照 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。
命名法の説明に関しては、「Quick Reference」を参照されたい。
- Srourら[2015]は、c.4690G>A(p.Ala1564Thr)は、選択的エクソン(エクソン40a)に生じるとしている。
CC2D2A
遺伝子構造
この遺伝子は38のエクソンから成り、1,620のアミノ酸から成るタンパク質をコードする。このタンパク質は、RPGRIP1Lがコードするタンパク質と共通のドメインを有する。
病的バリアント
この遺伝子の病的バリアントは、Meckel症候群やJSRDを引き起こすと同時に、COACH症候群のバリアントでもある。ヌルバリアントは、より重度型(多くは致死性)のMeckel症候群の表現型となって現れる[Mougou-Zerelliら2009]。フランス系カナダ人において、CC2D2Aのいくつかの病的バリアントで、創始者効果と思われるバリアントが同定されている[Srourら2012b,Srourら2015]。
正常遺伝子産物
このタンパク質は、コイルドコイルドメインとC2カルシウム結合ドメインをもち、繊毛発生に決定的に重要な役割を果たすと考えられている。選択的スプライシングにより、複数の転写産物のバリアントが生じる。CC2D2Aは基底小体に局在し、CEP290と物理的相互作用を行う[Gordenら2008]。
異常遺伝子産物
CC2D2Aタンパク質が失われることで、ヒトの疾病につながる。
ゼブラフィッシュホモログの機能喪失により、前腎の嚢胞(腎嚢胞に相当)、ならびに、繊毛機能障害に一致したその他の変化が生じる[Gordenら2008]。
CEP290
遺伝子構造
この遺伝子は54のエクソンから成り、ゲノムDNA上で93.2kbのサイズを有する。全エクソンを含む転写物のサイズは7,972bpである。選択的スプライシングにより、数種類のアイソフォームが生じる。
病的バリアント
CEP290には100種類を超える病的バリアントが同定されており、その大多数はトランケーションが予測されるもの(全部で112種類、そのうち40がナンセンスバリアント、48がフレームシフトバリアント)である。JSに関連するものとして、大きな部分欠失のヘテロ接合も1つ報告されてはいるものの、大多数のトランケーションバリアントは、小さな挿入あるいは欠失によって生じたものである。ミスセンスバリアントは3つだけである。スプライシングに影響を及ぼすものが20存在する[Coppietersら2010]。
CEP290の病的バリアントに伴って生じる表現型のスペクトラムの幅は広く、LCA、ネフロン癆、Senior-Løken症候群、JS、Meckel症候群、Bardet-Biedl症候群などが含まれる(表3参照)。遺伝型-表現型相関を明確に述べることは難しいが、いくつかの相関が報告されており、それらはlosus-specific database CEP290baseにまとめられている[Coppietersら2010]。
正常遺伝子産物
CEP290は、290kdの中心体タンパク質(nephrocystin-6とも呼ばれる)をコードする。
このタンパク質は、2,479のアミノ酸残基から成る。Nephrocystin-6は、腎嚢胞の形成に関与する転写因子であるATF4の活性を調節することが知られている中心体タンパク質である。このタンパク質には、推定13のコイルドコイルドメイン、SMC(染色体構造維持;structural maintenance of chromosomes)ATPase相同領域、6つのKIDモチーフ、3つのトロポミオシン相同ドメイン、1つのATP/GTP結合部位モチーフAが含まれる。このタンパク質は、中心体と繊毛に局在し、N-グリコシル化、チロシン硫酸化、リン酸化、N-ミリストイル化、アミド化関連部位を有する。Nephrocystin-6は、CC2D2Aやmeckelinをはじめとする他のJSRD関連タンパク質と相互作用することがわかっている[Gordenら2008,Leitchら2008,Tallilaら2008]。
異常遺伝子産物
CEP290の機能喪失により疾患が生じる。ゼブラフィッシュのノックダウン実験では、小脳、腎、網膜の発生異常が確認されている[Sayerら2006]。データからは、胚発生中のマウスの小脳で、このタンパク質が発現していることが示唆されている[Valenteら2006b]。
cep290に病的バリアントを有する自然発生の2つの動物モデルがrd16マウスとアビシニアン猫で確認されており、どちらも進行性の網膜変性を示すものの、腎や小脳の異常はみられない[Coppietersら2010]。
CSPP1
遺伝子構造
CSPP1は、876のアミノ酸から成る短いアイソフォームと、1,221のアミノ酸から成る長いアイソフォームをコードしている[Patzkeら2006,Tuzら2014]。
病的バリアント
報告されている病的バリアントの大部分をトランケーション型ナンセンスバリアント、トランケーション型フレームシフトバリアント、スプライス部位バリアントが占め、変異部位はタンパク質全体に分布している[Akizuら2014,Tuzら2014]。また、スプライシングの異常と、下流におけるフレームシフトを誘導する1つのミスセンスバリアントも報告されている[Tuzら2014]。この遺伝子の病的バリアントを有する罹患者においては非常に多様な表現型が出現するが、そうしたものを説明できるだけの明確な遺伝型-表現型相関は存在しない模様である。
表5:こので取り上げたCSPP1の病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.363_364delTA | p.His121GlnfsTer22 | NM_024790.6 NP_079066.5 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。
GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。
命名法の説明に関しては、「Quick Reference」を参照されたい。
正常遺伝子産物
この遺伝子のコードする中心体紡錘体極関連タンパク質1(centrosome spindle pole-associated protein 1;CSPP1)は101.5kd(長いアイソフォームは142kd)で、5つのコイルドコイルドメインを有する。このタンパク質は、中心体と相互作用することで、細胞周期の進行、有糸分裂の際の紡錘体の構成に一定の役割を果たす[Patzkeら2006]。
異常遺伝子産物
CSPP1関連JS罹患者の線維芽細胞では、繊毛が少ない、ないし短いという繊毛発生障害がみられる[Tuzら2014]。ソニックヘッジホッグシグナル伝達経路の障害の報告もみられる[Shaheenら2014]。
INPP5E
遺伝子構造
INPP5Eは、9つのエクソン、3,440bpのmRNAから成り、644のアミノ酸から成るタンパク質をコードする。
病的バリアント
この遺伝子の触媒活性ホスファターゼドメイン内に生じるミスセンスバリアントにより、いくつかのタイプのJSRDが引き起こされる[Bielasら2009]。Bardet-Biedl症候群様MORM症候群の1家系で同定された病的バリアントは、タンパク質の未成熟トランケーション、末端の18アミノ酸の欠失をもたらすものである[Jacobyら2009]。
正常遺伝子産物
この遺伝子にコードされるタンパク質は、72kdのイノシトールポリリン酸5ホスファターゼ(イノシトール1,4,5三リン酸[InsP3]5ホスファターゼとも呼ばれる)である。これは、細胞内カルシウムを動員すること、ならびに、種々の刺激に対する細胞応答を仲介するセカンドメッセンジャーとして働くことで、ホスファチジルイノシトールのシグナル伝達に関与する酵素である。この酵素は、一次繊毛の中心核に局在し、ホスファチジルイノシトールの代謝や安定性に影響を及ぼしている模様である[Jacobyら2009]。
異常遺伝子産物
JS関連病的バリアントにより、この酵素の5ホスファターゼ活性が損なわれ、繊毛のホスファチジルイノシトール比に変化が生じ、繊毛の安定性が低下する。オーソログ遺伝子のホモ接合性欠失を有するマウスは、無眼球、多指趾、嚢胞腎、骨格異常、口蓋裂、脱出脳症のような脳奇形を呈して、出生後ほどなく死亡する[Jacobyら2009]。末端の18のアミノ酸の欠失は、繊毛内におけるこのタンパク質の局在に影響を及ぼしている模様である[Jacobyら2009]。
KIAA0586
遺伝子構造
KIAA0586(TALPID3)は、34のエクソンをもち、最長のアイソフォームで1,644のアミノ酸から成るタンパク質をコードしている。少なくとも6つのアイソフォームが報告されている[Roosingら2015]。
病的バリアント
症候は幅広いスペクトラムをもって現れるが、これをもたらす病的バリアントとしては、通常はトランケーションバリアント、そして時にミスセンスバリアントがある[Albyら2015,Bachmann-Gagescuら2015b,Malicdanら2015,Roosingら2015]。
比較的多くみられる病的バリアントであるc.428delGは、一般集団において300人に1人の割合で生じると予測されているが[Roosingら2015]、いくつかのコホートでは、それに続く第2の病的と思われるバリアントは、まだ同定されるに至っていない[Bachmann-Gagescuら2015b,Roosingら2015]。
注目すべきことは、早期終止のタンパクが形成されるKIAA0586の反復性マルチエクソン欠失がMalicdanら[2015]により同定されたが、こうした遺伝子内大規模欠失が、他の研究グループの報告したコホートにおいて果たして評価の対象となっていたかどうか明らかでないことである。
表6:このGeneReviewで取り上げたKIAA0586の病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.428delG | p.Arg143LysfsTer4 | NM_01244189.1 NP_001231118.1 |
| del exon8-exon10 | NM_001244189.1 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。
GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。
命名法の説明に関しては、「Quick Reference」を参照されたい。
正常遺伝子産物
KIAA0586は、4つのコイルドコイルドメイン、1つのC末端プロリンリッチドメインをもつと予測される中心体タンパク質をコードしている。これは、繊毛の発生やヘッジホッグシグナル伝達において必要である。ニワトリにおけるオーソログタンパク質はTALPID3であるが、これは、発生途上にあるニワトリの四肢・神経管・体節におけるソニックヘッジホッグの伝達に不可欠なものである[Malicdanら2015]。
異常遺伝子産物
KIAA0586は、中心体の遠位端部の環状構造の組立てにおいて補助的役割を果たすことで、繊毛へのタンパク質輸送を仲介する。KIAA0586が欠損することで、繊毛小胞(ciliary vesicle)の形成、ならびに中心体の移動障害が生じる。ニワトリ胚、変異マウス、ゼブラフィッシュ胚においては、KIAA0586が正常に発現しないと、一次繊毛を欠く細胞が形成され、顔面・四肢・神経管の障害が引き起こされる[Malicdanら2015]。ある報告の罹患者群では、KIAA0586病的バリアントは、すべて、中心体の局在に必要な高度に保存されたドメインよりも前の部分に生じていた[Malicdanら2015]。さらに、KIAA0586の病的バリアントを有する罹患者由来の線維芽細胞は、繊毛形成の低下、繊毛が存在した場合でもその長さの縮小、ソニックヘッジホッグシグナル伝達の変化といったものを呈した[Albyら2015,Malicdanら2015]。
MKS1
遺伝子構造
MKS1は21,170bpの長さで、18のエクソンをもち、559のアミノ酸から成るタンパク質をコードしている。この遺伝子では、複数の転写バリアントが複数のアイソフォームをコードする。
病的バリアント
この遺伝子には、ミスセンスバリアント、ナンセンスバリアント、トランケーションバリアントなど、さまざまなものが報告されている。そしてその中の1つに、本遺伝子の病的バリアントに起因するJS罹患者11人中4人にみられた反復性のバリアント(p.Ser372del)がある[Romaniら2014,Slaatsら2016]。より重度のMeckel症候群の表現型を示す罹患者については、JS罹患者の場合より、MKS1のバリアントがより重大なものであることが予測される。JS罹患者の場合は、この遺伝子におけるバリアントのうちの少なくとも1つは、非トランケーション型であることがふつうである。
表7:このGeneReviewで取り上げたMKS1の病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.1115_1117delCCT | p.Ser372del | NM_017777.3 NP_060247.2 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。
GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。
命名法の説明に関しては、「Quick Reference」を参照されたい。
正常遺伝子産物
この遺伝子によりコードされるタンパク質は、基底小体のtransition zoneに局在し、繊毛細胞における一次繊毛の形成に必要なものである。この遺伝子により重度のバリアントが生じた場合は、Meckel症候群1型やBardet-Biedl症候群13型となって現れる。
異常遺伝子産物
Slaatsら[2016]は、この遺伝子の病的バリアントを有するJS罹患者の線維芽細胞を調べ、細胞の繊毛の数自体は変わらないか、もしくは減少しており、対照群に比べ、その長さには大きなばらつきの幅がみられたことを報告している。それに加え、重要な繊毛タンパク質であるARL13BとINPP5Eの分布にも変化がみられたとし、通常であれば、INPP5Eは機能的transition zoneを必要としつつARL13B依存性に繊毛に沿う形で分布するが、分析を行った罹患者群ではこのtransition zoneが欠如しているようであるとしている[Slaatsら2016]。
NPHP1
遺伝子構造
NPHP1は20のエクソンから成り、そのcDNAは3,713bpである。この遺伝子は、2つの大きな逆位反復エレメントに挟まれた領域に位置し、nephrocystin-1をコードしている。
病的バリアント
NPHP1から隣接遺伝子BENEの一部にまたがる約290kbの欠失のホモ接合[Saunierら2000,Parisiら2004a]に加え、NPHP1の1塩基バリアントも時に同定されることがある[Hoefeleら2005](詳細は表Aを参照)。家族性若年性ネフロン癆1型あるいはSenior-Løken症候群よりも重度の表現型を呈する例の中には、NPHP1のホモ接合性欠失に加えてAHI1あるいはCEP290のヘテロ接合性の変化を有するものがあることから、複数のmodifier遺伝子の関与が示唆されている[Toryら2007]。
正常遺伝子産物
Nephrocystin-1は、733のアミノ酸から成るタンパク質で、他のタンパク質との相互作用を仲介する働きを担うと考えられるsrc相同ドメイン3(SH3)をもつ。Nephrocystinは、細胞の一次繊毛、細胞間結合部、基底小体に局在し、その場所で、細胞分裂の調節や、細胞間・細胞-マトリックス間接着のシグナル伝達に一定の機能を果たしている模様である[Hildebrandtら2009]。NephrocystinはAHI1をはじめ、INVS、NPHP3、NPHP4などのタンパク質とも相互作用する。なお、後3者は、別のタイプのネフロン癆で変異が生じる遺伝子がコードするタンパク質である。
異常遺伝子産物
Nephrocystinは、その他数多くの繊毛タンパク質と関連し合うこと、ならびに、腎上皮の繊毛/基底小体に局在することが知られていることから、尿細管の発生に決定的に重要な役割を果たしているものと考えられている。
RPGRIP1L
遺伝子構造
この遺伝子は、3,948bpで26のエクソンをもち、1,315のアミノ酸から成るタンパク質をコードしている。
病的バリアント
ミスセンスバリアント、ナンセンスバリアント、スプライス部位バリアントなど、さまざまな種類のものが同定されている。一般に、より重度のトランケーションバリアントでは致死性のMeckel症候群の表現型が生じ、より軽度型のバリアントではCOACH型の表現型を含むJSRDが生じる[Delousら2007,Wolfら2007]。さらに、p.Ala229Thrのバリアントは、他の遺伝子の病的バリアントに起因して繊毛病に至った罹患者における網膜変性の発生に関与している[Khannaら2009]。
表8:このGeneReviewで取り上げたRPGRIP1Lの病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.685G>A | p.Ala229Thr | NM_015272.2 NP_056087.2 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。
GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。
命名法の説明に関しては、「Quick Reference」を参照されたい。
正常遺伝子産物
この遺伝子のコードするタンパク質(protein fantom)は、複数のコイルドコイルドメイン、1つのC2カルシウム結合ドメイン、1つのRPGR(網膜色素変性GTPアーゼ調節因子;retinitis pigmentosa GTPase regulator)相互作用ドメイン、1つの中心体タンパク質関連ドメインを有している。このタンパク質は、基底小体-中心体複合体、あるいは繊毛細胞の一次繊毛や中心体に局在し、nephrocystin-4と相互作用する。なお、nephrocystin-4というのは、ネフロン癆のいくつかのタイプ、ならびにSenior-Løken症候群において障害を受けるタンパク質である[Artsら2007]。RPGRIP1Lには、それぞれ別のタンパク質アイソフォームをコードする2つの転写バリアントが同定されている。
異常遺伝子産物
RPGRIP1Lの機能喪失により、疾患が生じる。また、p.Ala229Thrの変異により、RPGRIP1Lのコードするタンパク質とRPGRタンパク質との相互作用に変化が生じ、視細胞の喪失につながる模様である[Khannaら2009]。
TCTN2
遺伝子構造
TCTN2(tectonic family member 2)は18のエクソンをもち、いくつかの転写産物をコードするが、最長のものは697のアミノ酸から成る。この遺伝子は、N末端のシグナルペプチド、ならびにC末端にある1つの膜貫通ドメインをコードしている。なお、後者はショウジョウバエのオーソログ中においても保存されている[Reiter & Skarnes 2006]。
病的バリアント
JSRDやMeckel症候群で、この遺伝子のナンセンスバリアント、フレームシフトバリアント、スプライス部位バリアントがみられる[Sangら2011,Shaheenら2011]。
正常遺伝子産物
Tectonic-2である。Tctn2は、マウスにおいてはヘッジホッグシグナル伝達ならびに繊毛発生の調節に関与していることがわかっている。Tctn2は、Mks1やCc2d2aと相互作用する。
異常遺伝子産物
TCTN2の機能喪失により、疾患が生じる。繊毛が基本単位の組立てにより形成されていること、ならびに、さまざまな細胞突起内でさまざまなタンパク質がそれぞれ異なった働きの相互作用をしていることを表す用語として、NPHP、JS、MKSといったタンパク質も含め、繊毛「インタラクトーム」という考え方が提唱されている[Sangら2011]。
TMEM67(MKS3)
遺伝子構造
この遺伝子は28のエクソンをもち、ゲノムDNA上で62.0kbの範囲にまたがり、全長の転写産物のサイズは3,467bpである[Smithら2006]。スプライスバリアントが少なくとも1つ存在し、これは29エクソン、3,280bpで、995残基から成るタンパク質をコードする[Ensembl Database]。
病的バリアント
Joubert症候群とその関連疾患の罹患者で同定されている病的バリアントには、異常転写産物を産生するスプライス部位バリアントと、ミスセンスバリアントがあり、両者とも、Meckel症候群を引き起こすような重度で致死性のバリアントではなく、軽度の表現型をもたらすhypomorphicなアレルであろうと思われる[Smithら2006,Baalaら2007]。この遺伝子の病的バリアントは、肝病変を伴うJS(COACHバリアント)の罹患者に多くみられる[Iannicelliら2010]。
正常遺伝子産物
Meckelinである。このタンパク質は、計算上の分子量が108kd、995のアミノ酸から成り、1つのシグナルペプチド、少なくとも2つのシステインリッチリピート、490アミノ酸から成る細胞外領域、その後、7つの膜貫通ドメイン、短い30残基の細胞質側末端といった構成であると予測されている[Smithら2006]。このタンパク質は、腎・胆道の上皮細胞その他の繊毛細胞の一次繊毛や細胞膜に局在し、MKS1タンパク質と相互作用することがわかっている。MKS1は、Meckel症候群で障害を受けるタンパク質である。Meckelinは、繊毛発生初期における中心体の細胞先端の表面への移動に関与し、繊毛の発生・機能の両面に不可欠なものである[Daweら2007]。
異常遺伝子産物
TMEM67の機能喪失により、疾患が生じる。TMEM67の1塩基バリアントを有する自然発生変異ラットwpk/wpkでは、多発性嚢胞腎、ならびに脳梁無形成を伴う水頭症を示す[Smithら]。自然発生欠失変異マウスでもこれと同様の表現型が確認され、多嚢胞性腎症により、通常、3週齢までに死亡する。一部、水頭症を発症する例もみられる[Cookら2009]。
TMEM216
遺伝子構造
TMEM216は6つのエクソンをもつ。最長のスプライスアイソフォーム(NM_001173990)は148のアミノ酸から成るタンパク質をコードする。TMEM216もTMEM138も繊毛遺伝子で、どちらもJSの原因遺伝子であるが、両者の中間領域の23kbが両遺伝子の発現を調節している模様である[Leeら2012b]。
病的バリアント
病的バリアントとしては、ミスセンスバリアント、ナンセンスバリアント、スプライス部位バリアントなどがある。多くみられるある1つのバリアント(c.218G>T)は、p.Arg73Leuというタンパク質の変化をきたすものであるが、これは、アシュケナージ系ユダヤ人集団にみられる創始者バリアントであるように見受けられ、その保因者頻度は、92人に1人から100人に1人である[Edvardsonら2010,Valenteら2010]。病的バリアントの多くは、タンパク質のトランケーションを引き起こすことが予測されるものであるが、こうしたバリアントは、致死性のMeckel症候群の表現型の原因ともなりうるものである[Valenteら2010]。
表9:このGeneReviewで取り上げたTMEM216の病的バリアント
| DNAヌクレオチドの変化 | 予測されるタンパク質の変化 | 参照配列 |
|---|---|---|
| c.218G>T | p.Arg73Leu | NM_001173990.2 NP_001167461.1 |
表中のバリアントは、著者の提供したものをそのまま載せたもので、GeneReviewsのスタッフが独立した立場でバリアントの分類を確認したものではない。
GeneReviewsは、Human Genome Variation Society(varnomen.hgvs.org)の標準命名規則に準じた表記を行っている。
命名法の説明に関しては、「Quick Reference」を参照されたい。
正常遺伝子産物
最長のアイソフォームは、膜貫通タンパク質216(transmembrane protein 216)で、4つの疎水性膜貫通ドメインを有する4回膜貫通タンパク質である。アイソフォームを含むこれら一群のタンパク質は、Wnt受容体をはじめとするパートナータンパク質のもつシグナル伝達や輸送に関する特性を調節しているものと思われる。TMEM216は、一次繊毛の基底部に局在し、meckelinと1つの複合体を形成する。このmeckelinは、同じく膜貫通タンパク質で、TMEM67によってコードされ、これが障害を受けることで同じくJSRDが生じるタンパク質である[Valenteら2010]。さらに、TMEM216とTMEM138は、それぞれ別の小胞プールに局在して、繊毛の組立てに必要なタンパク質をゴルジ体から一次繊毛へと運搬するという点で、繊毛発生に必要な存在である[Leeら2012b]。
異常遺伝子産物
ゼブラフィッシュでtmem216に障害を加えると、原腸陥入の障害をはじめとする、繊毛機能の変化に特徴的なさまざまな病変が引き起こされる[Valenteら2010]。
表1bにある各遺伝子に関する情報は、こちらをクリック(本稿末尾)
更新履歴:
- Gene Review著者: Melissa Parisi, MD, PhD, Ian Glass, MD
日本語訳者: 末國久美子、川目裕
(お茶の水女子大学大学院人間文化創成科学研究科ライフサイエンス専攻遺伝カウンセリング領域 )
Gene Review 最終更新日: 2007.3.8.日本語訳最終更新日: 2011.8.18. -
Gene Reviews著者: Melissa Parisi, MD, PhD and Ian Glass, MD.
日本語訳者: 佐藤康守(たい矯正歯科)、櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日:2017.6.29. 日本語訳最終更新日: 2023.4.2.[in present]
原文: Joubert Syndrome
![]()