白質ジストロフィー概説
(Leukodystrophy Overview)
Gene Reviews著者: Adeline Vanderver,MD, Davide Tonduti, MD, Raphael Schiffmann, MD, Johanna Schmidt, MPH, MGC, CGC, and Marjo S van der Knaap, MD, PhD.
日本語訳者: 吉田誠克(京都府立医科大学 神経内科)
Gene Reviews 最終更新日: 2014.02.06.日本語訳最終更新日: 2017.03.24
要約
疾患の特徴
白質ジストロフィーは中枢神経系の白質が障害される遺伝性の髄鞘疾患であり,末梢神経系の髄鞘は障害される場合とされない場合がある.白質路の障害はほぼ例外なく運動障害をもたらし,幼児期早期に筋緊張低下として現れ,経過とともに痙性へと進行する.この運動障害は軽度の痙性対麻痺から随意運動を制限する重度の痙性四肢麻痺まで様々である.さらに,運動機能障害は嚥下,咀嚼および(症例により)呼吸といった生命機能を著しく障害する可能性が高い.疾患により異なる他の所見として錐体外路性運動異常(ジストニアやジスキネジア),失調,けいれん,認知発達の遅延や認知機能変化が挙げられる.
診断・検査
各症例がどの白質ジストロフィーに該当するかを確立する方法として以下が挙げられる.
- 病歴聴取と詳細な家族歴
- 身体所見と神経学的所見
- 頭部MRIの検討
- 白質のT2強調画像高信号は白質ジストロフィーの診断に必要なMRI所見である.
- T1強調画像所見は様々である:等信号あるいは高信号は髄鞘形成不全性白質ジストロフィーに合致する;低信号は脱髄性白質ジストロフィーに合致する.
- 特異的な臨床検査を行う際には,しばしば分子遺伝学的検査が含まれる(1種類ずつ遺伝子検査を行うか,白質ジストロフィーを標的にしたマルチ遺伝子パネルを利用するか)
臨床的マネジメント
対症療法:
治療は症状に対するもので,原則,白質ジストロフィー患者のケアの経験が豊富な専門家による多職種で行われる.薬物治療として筋緊張の調整や筋に対する神経伝達をブロックする(化学的除神経)薬剤が使われてきた.集中的な理学療法は動作や機能の改善を目的に使用されてきた.ジストニアとジスキネジアの薬理学治療は有意な機能改善をもたらす可能性がある.失調,けいれん,認知の問題に対する治療は個々の必要性に応じて標準の方法で行われる.
原疾患の症状予防:
疾患初期に造血幹細胞移植(HSCT)あるいは骨髄移植(BMT)を施行することにより初期徴候の防止が可能な白質ジストロフィーがある.
サーベイランス:
成長や栄養状態の定期評価;整形外科合併症に対する理学所見や股関節および脊椎のX線写真;けいれんの再発や徴候に関する病歴聴取.
避けるべき薬剤/環境:
軽度の頭部外傷や感染は疾患を増悪させる可能性がある.
初期予防(HSCTあるいはBMTなど)が可能な白質ジストロフィーであれば,早期診断・早期治療が有益と考えられる無症状のリスクのある近親者に対して検査を行うことは適切である.
遺伝カウンセリング
遺伝的要因を特定できる白質ジストロフィーは常染色体優性遺伝形式,常染色体劣性遺伝形式,あるいはX連鎖性劣性遺伝形式にて遺伝しうる.家族のリスクに関する遺伝カウンセリングは正確な診断,各家系における遺伝形式の決定,分子遺伝学的検査の結果に基づく.リスクの高い妊娠に対する出生前診断はいくつかの白質ジストロフィーでは可能だが,それは家系内の病原性バリアントが明らかな場合である.多くの白質ジストロフィーでは未だ遺伝学的要因が特定されていない;遺伝学的要因が特定されても,別の遺伝様式が生じうる.
白質ジストロフィーの定義
白質ジストロフィーという用語は,白質形成不全症,脱髄,白質脳症といった関連用語と同様に,広範な疾患群に適用されている.
このGeneReviewの章では,以下の定義を用いる:白質ジストロフィーは中枢神経系の白質が障害される遺伝性の髄鞘疾患であり,末梢神経系の髄鞘は障害される場合とされない場合がある.
白質ジストロフィーは以下の所見を共有する:
- グリア細胞あるいは髄鞘の異常,その神経病理はオリゴデンドロサイト,アストロサイト,その他の非ニューロンの一次性障害により特徴づけられる.
- MRI画像
- 白質のT2強調画像高信号の存在が必須.
- T1強調画像所見は様々:等信号あるいは高信号は髄鞘形成不全性白質ジストロフィーに合致;低信号は脱髄性白質ジストロフィーに合致.
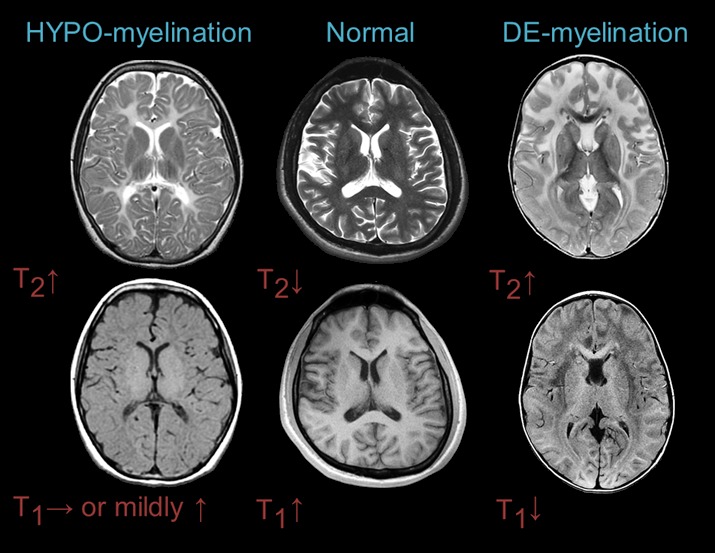
図1.
髄鞘形成不全性白質ジストロフィーは病変白質部位においてT2強調画像にて高信号(↑),T1強調画像にて等信号(→)あるいは高信号(↑)を示す.脱髄性白質ジストロフィーは病変白質部位においてT2強調画像にて高信号(↑),T1強調画像にて低信号(↓)を示す.
白質ジストロフィーの臨床症状
このGeneReviewの章で用いた定義を満たす多くの白質ジストロフィーを図1に挙げている.
白質路の障害によりほぼ例外なく小児期早期には筋緊張低下として現れる運動障害が引き起こされ,時間の経過とともに痙性に進展する.運動障害の程度は軽度の痙性対麻痺から随意運動が制限される重度の痙性四肢麻痺まで様々である.さらに,運動機能障害は嚥下,咀嚼,(時には)呼吸といった生命機能を著しく障害する可能性が高い.痙性は側弯や大関節脱臼などの整形外科的合併症の原因となりうる.
著しい錐体路障害(痙性など)はジストニアやジスキネジアなどの錐体外路性運動障害の存在を時に隠す,あるいは分かりにくくする可能性がある.例えば,MCT-8特異的甲状腺ホルモン細胞輸送体欠損症ではジストニアが重要な所見である.失調はいくつかの白質脳症において顕著な所見であり身体の不自由を生じうる;例えば,中枢神経系白質形成不全症を伴う小児失調症/白質消失病(CACH/VWM)や歯牙低形成および低ゴナドトロピン性性腺機能低下を伴う髄鞘形成不全症(4H症候群)が挙げられる.
けいれんはしばしば白質ジストロフィーの晩期に出現する症候であるが,アレキサンダー病のように初期の症候として出現する白質ジストロフィーもまれに存在する.
認知発達の遅延あるいは経時的な認知機能変化は,運動機能低下と比較するとはるかに目立たないが,白質ジストロフィーの小児あるいは成人において一般的に認められる.認知機能の進行性喪失は白質ジストロフィーの大多数において緩徐であるため,認知症は早期の症候とはならない.
白質ジストロフィーの有病率
白質ジストロフィーの頻度に関する疫学データは全体的に限られている[Heim et al 1997, Bonkowsky et al 2010]. さらに,これらの研究から特定の白質ジストロフィーの相対頻度を確実に推定することは困難である.
専門病院や一般の小児神経科で定期的に診察されている白質ジストロフィーにおいてはより有用な有病率が得られている;アレキサンダー病[van der Knaap et al 1999], X連鎖性副腎白質ジストロフィー[Bezman et al 2001], 異染性白質ジストロフィーが該当する.
ある種の成人発症のみの白質ジストロフィーや髄鞘形成不全性白質ジストロフィーはより定義づけされてきているので,遺伝性白質疾患の異質性は徐々に認識されてきている.
白質疾患患者のおよそ50%はその異質性や複雑性のため特定の病因が不明であることを認識しておくことは重要である[Schiffmann & van Knaap 2009].
白質ジストロフィーの鑑別診断
白質ジストロフィーではないが顕著な白質病変を伴う遺伝性疾患を表2(pdf)に列挙した;以下のものが含まれる:
- 遺伝性血管症は,CADASIL, COL4A1およびCOL4A2関連疾患などの多巣性の白質異常をもたらす小血管壁の遺伝的異常に関連し,白質信号異常をきたしうる.末期には脳室周囲および深部白質に融合した信号異常を認めるが,典型的には中心灰白質構造や脳幹に多巣性の異常を伴う.これらは一次性のグリア細胞疾患ではないため,白質ジストロフィーとはみなされない.
- 大脳皮質あるいは他の灰白質構造内の一次性ニューロン障害を伴う中枢神経疾患(これらにおいても脳MRIは著しい白質異常を示す).典型的な所見は重篤な脳症やけいれんである.
- GM1ガングリオシドーシス,GM2ガングリオシドーシス,神経セロイドリポフスチノーシスのような疾患の乳児型は,一次性にニューロンを侵すとともに髄鞘化の正常な過程も崩壊させるため,顕著な白質異常を伴うと考えられる.これらの疾患の晩期発症型では最終的に大脳白質を障害する;軽度の信号異常は軸索と髄鞘の喪失を伴うワーラー変性に関連する.
- MCT-8特異的甲状腺ホルモン細胞輸送体欠損症のような疾患では,髄鞘化は極端に遅延するが,経過とともに発展して最終的にはほとんど完成するようになる.
- 先天代謝異常症:有機酸血症やアミノ酸代謝異常症,その他多くの疾患にみられる著しい二次性白質異常.
- 白質と灰白質の両者を障害する疾患.白質損傷と灰白質病変の両方をきたす例としてはMELASのようなミトコンドリア病,POLG関連疾患,家族性血球貪食症候群などが挙げられる.
著しい白質病変を伴う他の疾患には以下が含まれる:
- 多発性硬化症や感染症,感染後,自己免疫の病態などの脱髄による後天性中枢神経系脱髄疾患.メンデル遺伝形式を示さないが,これらの疾患の中には遺伝的素因を含む多因子の病態を有するものがあるかもしれない.
- 急性散在性脳脊髄炎,多発性硬化症,視神経脊髄炎のような疾患は突然発症や多相性の経過により遺伝性白質疾患とは異なる.脳MRI異常も経時的に多様な変化あるいは時には改善を示しながら,また治療を行うことにより,多巣性でまばらになる傾向がある[Schiffmann & van der Knaap 2009].
- 髄鞘の中毒による損傷.ヘロイン中毒あるいはメソトレキセート関連毒性において認められる.
- 中枢神経系損傷.特に周産期に生じ,典型的には不規則で著しい白質信号異常となり,白質容量の喪失に至る可能性がある.
白質ジストロフィー患者に対する評価方法
白質ジストロフィーを疑った場合は,以下のアプローチが特定の白質ジストロフィーを決定するのに有用で,予後や遺伝カウンセリングを検討する助けとなる.
特定の白質ジストロフィーの確立は病歴と詳細な家族歴を得て,身体診察と神経診察を行い,脳MRIを精査し,分子遺伝学的検査を含む特異的な生化学検査を行う.
注:ここで述べたとおりの診断アプローチを施行したにもかかわらず,大多数の白質ジストロフィー患者においては臨床的に(研究ではなく)特定の診断を確立することはできない[van der Knaap et al. 1999, Schiffmann & van der Knaap 2009].
病歴
臨床症状の病歴は特定の白質ジストロフィーを同定するのに有用である;しかしながら,多くの症例で非特異的な機能喪失(主に運動)しか認めず,病歴のみでは特異的診断への洞察は提供されない.
髄鞘形成不全性白質ジストロフィーにおいて,助けとなる診断手掛かりは以下の例が挙げられる:
- 先天性白内障:髄鞘形成不全と先天性白内障(HCC)
- 歯牙低形成および低ゴナドトロピン性性腺機能低下:POLR3-関連性白質ジストロフィー
- 重篤なけいれん:シアル酸蓄積症
脱髄性白質ジストロフィーにおいて,助けとなる診断手掛かりは以下の例が挙げられる:
- 繰り返す嘔吐:アレキサンダー病
- 副腎機能不全:X連鎖性副腎白質ジストロフィー(X-ALD)
- 期発症の自律神経機能不全:常染色体優性遺伝性成人発症白質ジストロフィー(ADLD)
- 慢性の脳脊髄液リンパ球増多あるいは(頻度が高いのは)反復性"無菌性髄膜炎":エカルディ・グティエール症候群 発熱あるいは転倒後の突然の機能喪失:中枢神経系髄鞘形成不全を伴う小児失調症/白質消失病(childhood ataxia
- with central nervous system hypomyelination/vanishing white matter)(および他の白質ジストロフィーの可能性もある)
家族歴
低緊張,痙性,ジストニア,けいれん,失調,認知発達の遅延や経時的な認知機能の変化をもつ人に焦点をあてた詳細な3世代の家族歴.
- ほとんどの白質ジストロフィーは常染色体劣性遺伝であるため,両親の近親婚や同胞の医学的問題に特別の注意が必要である.
- 親族の評価や病歴の再考が必要な場合がある.
身体診察と神経学的診察
ほとんどの場合,身体診察にて特異的な診断を示唆する所見は得られない;しかしながら,所見の中には特定の根底にある病態を示唆するものがある:
- 大頭症;表6参照(pdf)
- 歯列異常:POLR3-関連白質ジストロフィーあるいは眼歯指異形成症(ODDD)
- 成人の口蓋ミオクローヌス:アレキサンダー病
- 黄色腫:脳腱黄色腫(CTX)
- 皮膚色素沈着の異常:X連鎖性副腎白質ジストロフィー(X-ALD)
- 魚鱗癬:シェーグレン‐ラルソン症候群
- 網膜血管異常:石灰化と嚢胞を伴う大脳網膜微小血管症(CRMCC)
- 網膜検査におけるチェリーレッド斑(cherry red spot):GM1およびGM2ガングリオシドーシス
脳MRI所見
ステップ1
脳MRI異常のパターンが髄鞘形成不全性白質ジストロフィーあるいは脱髄性白質ジストロフィーのいずれかに合致するか明らかにする(図1)[van der Knaap et al 1999, van der Knaap & Valk 2005, Schiffmann & van der Knaap 2009].
ステップ2
髄鞘形成不全性あるいは脱髄性白質ジストロフィーであれば,病変のパターンが特定の診断を示唆するかどうかを決める.
髄鞘形成不全性白質ジストロフィー.神経画像における特異的なパターンは以下のとおりである(図2):
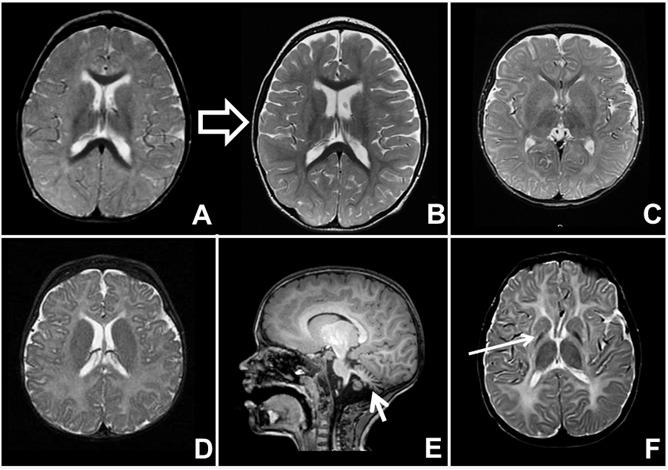
図2.
髄鞘形成不全性白質ジストロフィー‐MRIのパターン
A→B. SLC16A2(MCT8)の病原性バリアントが証明されたMCT8特異的甲状腺ホルモン細胞輸送体欠損症の男性における経時的な髄鞘化の改善:2歳時(A)と4.5歳時(B)
C. 白質容量の早期喪失を伴うGM1ガングリオシドーシス
D. ペリツェウス-メルツバッハー病の学童期の小児に認められた永続的で高度の髄鞘形成不全
E. POLR3関連白質ジストロフィー(4H症候群)に認められる小脳萎縮:矢印が萎縮
F. H-ABC症候群に認められた基底核病変:矢印が被殻の萎縮で,本疾患においては古典的な所見
- 経時的な髄鞘化の改善(例:MCT8特異的甲状腺ホルモン細胞輸送体欠損症;図2A→B参照)
- 皮質灰白質の高度萎縮(例:一次性ニューロン疾患や一部の古典的白質ジストロフィーにおいて様々にみられる;図2C)
- 皮質灰白質の萎縮を伴わない永続的な髄鞘形成不全(例:ペリツェウス‐メルツバッハー病;図2D)
- 小脳萎縮(例:POLR3関連白質ジストロフィー[4H症候群]; 図2E)
- 基底核異常(例:HABC症候群;図2F)
脱髄性白質ジストロフィー
- 白質異常が目立つ領域を同定する.
- 融合性病変か多巣性病変かを区別する(図3):
- 融合性の白質病変は必ずしも完全に対称性ではないが,脳のかなりの部分において白質異常が拡大し,しばしば特定の領域や伝導路を障害する.
- 多巣性の白質病変はより不連続で,しばしば非対称性で限定された領域の障害である.多巣性病変には特異的な鑑別診断がある.
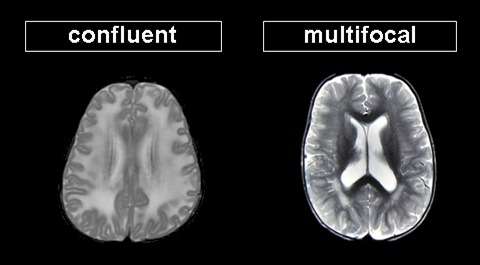
図3.
融合性の白質病変は必ずしも完全に対称性ではないが,脳のかなりの部分において白質異常が拡大し,しばしば特定の領域や伝導路を障害する.多巣性の白質病変はより不連続で,しばしば非対称性で限定された領域の障害である.多巣性病変には特異的な鑑別診断がある.
融合性疾患における神経画像の特異的パターンは以下のとおりである(Figure 4)
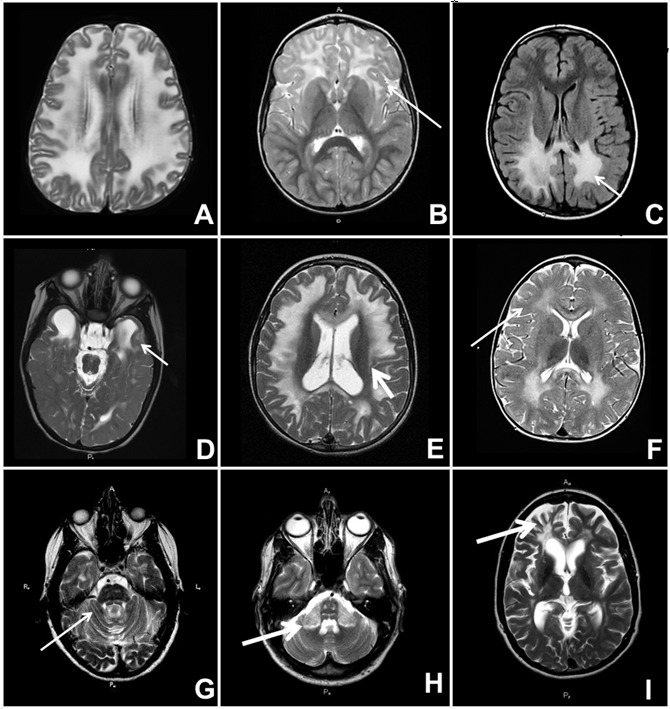
図4.
脱髄性白質ジストロフィー-MRIのパターン
A. 皮質下嚢胞をもつ大頭型白質脳症(megalencephalic leukodystrophy with subcortical cysts)患者におけるびまん性大脳障害
B. アレキサンダー病の小児における前頭葉優位病変:矢印が前頭領域の白質異常を示す
C. X連鎖性副腎白質ジストロフィーの小児における頭頂後頭葉優位病変:矢印が頭頂後頭領域の白質異常を示す
D. エカルディ・グティエール症候群における側頭葉優位病変:矢印が側頭領域の白質異常を示す
E. カーンズ・セイヤー症候群における皮質下優位病変:矢印は脳室周囲領域白質病変の回避を示す
F. 異染性白質ジストロフィーにおける脳室周囲優位病変:皮質下線維は回避されている:矢印は皮質下白質病変の回避を示す
G. 不均一な脳幹病変を伴う成人ポリグルコサン小体病患者における脳幹優位病変:矢印は脳幹の白質異常を示す
H. 常染色体優性成人発症白質ジストロフィー(ADLD)患者における小脳および中小脳脚優位病変;矢印は中小脳脚における白質異常を示す
I. 軸索スフェロイドをもつ遺伝性びまん性白質脳症(HDLS)患者における巨大な非対称性病変;矢印は前頭部領域の非対称性白質異常を示す
- びまん性の大脳障害(例:皮質下嚢胞をもつ大頭型白質脳症;図4A)
- 前頭葉病変(例:アレキサンダー病;図4B)
- 頭頂後頭葉病変(例:X連鎖性副腎白質ジストロフィー;図4C)
- 側頭葉病変(例:エカルディ・グティエール症候群;図4D)
- 皮質下病変(例:カーンズ・セイヤー症候群;図4E)
- 脳室周囲病変(例:異染性白質ジストロフィー;図4F)
- 脳幹病変(例:不均一な脳幹病変を伴う成人ポリグルコサン小体病;図4G)
- 小脳および中小脳脚優位病変(例:中小脳脚病変を伴う常染色体優性成人発症白質ジストロフィー(ADLD);図4H)
- 巨大な非対称性病変(例:軸索スフェロイドをもつ遺伝性びまん性白質脳症(HDLS);図4I)
診断アルゴリズム.MRI所見に基づくアルゴリズムについては,図5(脱髄および他の状態)と図6(髄鞘低形成)[Schiffmann & van der Knaap 2009].
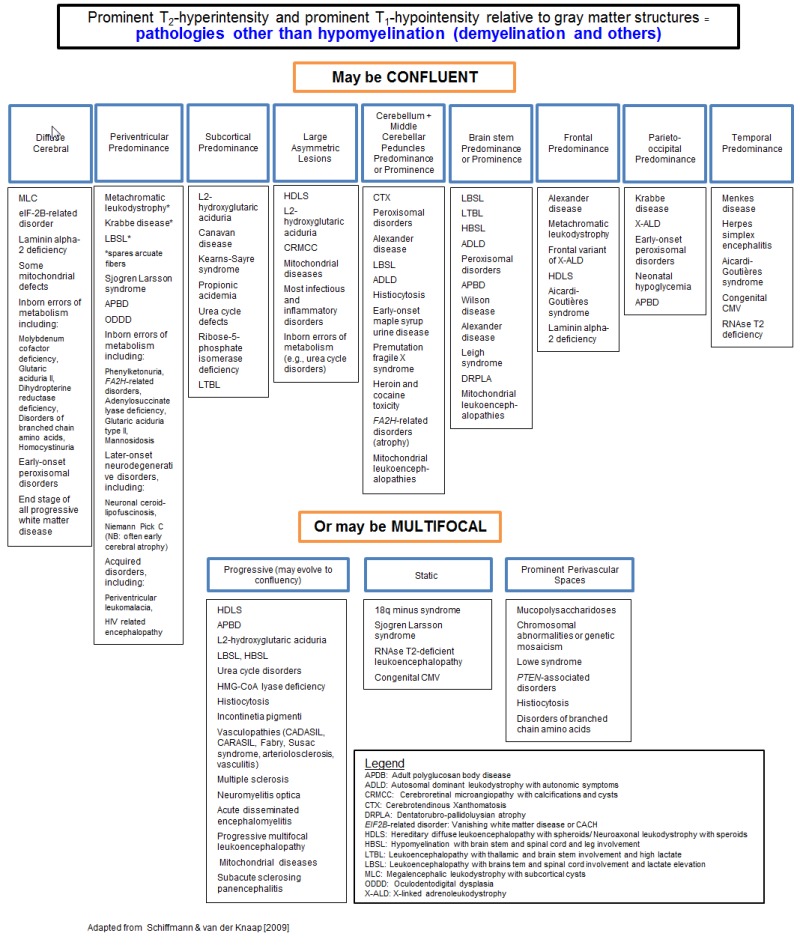
アルゴリズム1:脱髄および他の状態.Schiffmann & van der Knaap [2009]から適用.
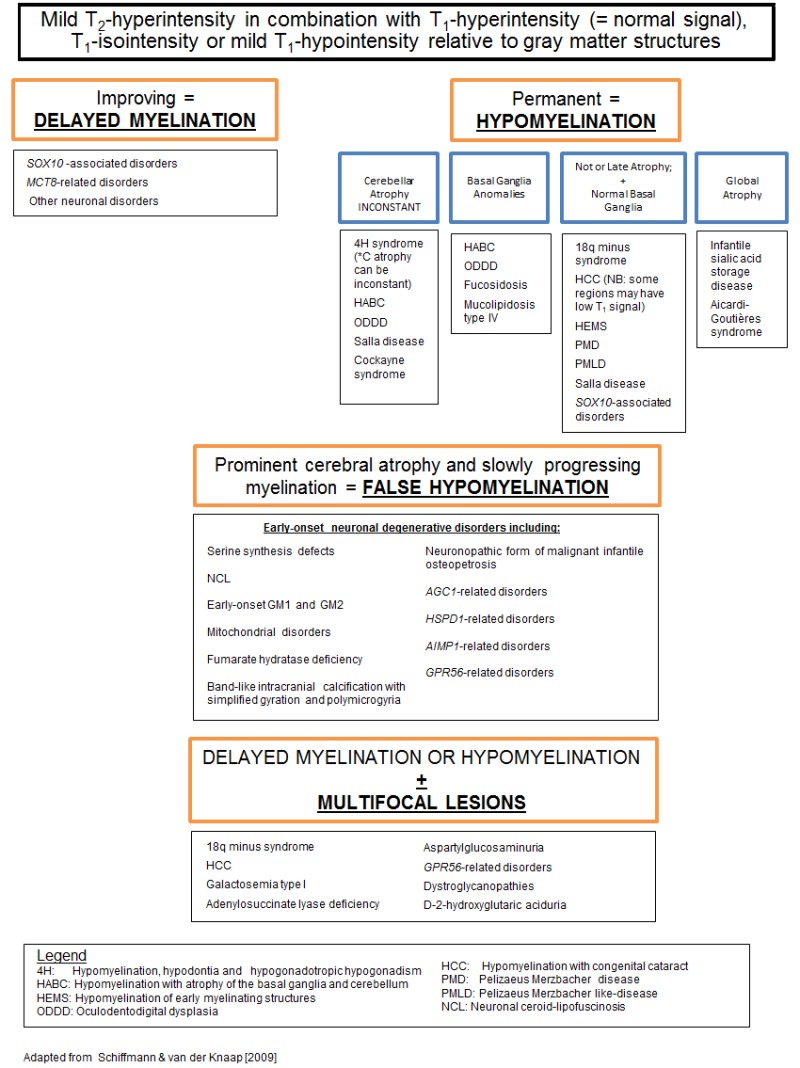
図6.
アルゴリズム2:髄鞘低形成.Schiffmann & van der Knaap [2009]から適用.
ステップ3
頭部MRI所見のパターンに加えて,特定の白質ジストロフィーを支持する次に示す関連徴候をみる:
- FLAIR画像にみられる白質の希薄化(rarefaction)と嚢胞(表3)(例:白質消失病(vanishing white matter disease):中枢神経系髄鞘形成不全を伴う小児失調症/白質消失(childhood ataxia with central nervous system hypomyelination/vanishing white matter); 図7A)
- MRIで容易には鑑別できないが,CTで可視化できるカルシウム沈着やヘモジデリン沈着(表4)(例:エカルディ・グティエール症候群;図7B参照)
- 異常白質内のT1強調画像における造影効果(表5)(例:X連鎖性副腎白質ジストロフィー;図7C参照)
- 大頭症を伴う白質ジストロフィー(表6)
- 皮質灰白質病変(表7)(例:POLG関連疾患;図7D参照)
- 歯状核にみられる小脳異常(表8)(例:L-2-ヒドロキシグルタル酸尿症;図7E参照)
- 脳梁の菲薄化(表9),特に膝部(例:遺伝性痙性対麻痺11;図7F参照)
- 非石灰化基底核病変(表10)(例:アレキサンダー病;図7G参照,基底核と前頭葉白質の信号異常)
- 脳幹病変(表11)(例:常染色体優性成人発症白質ジストロフィー(ADLD);図7H)
- 脊髄病変(表12)(例:LBSLを含む多くの疾患でみられる;図7I,脊髄内の伝導路障害の例)
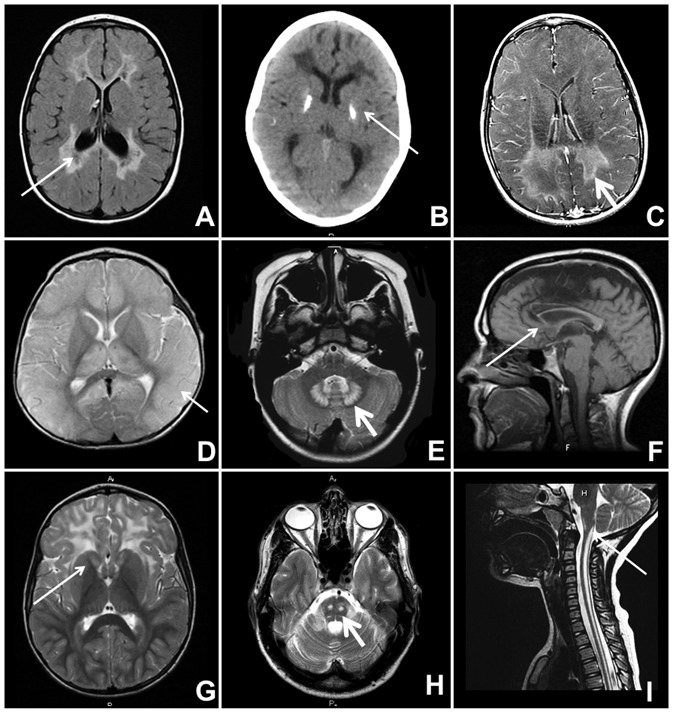
図7.
特異的な白質ジストロフィーに関連する画像
A. 白質消失病(vanishing white matter disease )のFLAIR画像にみられる白質の希薄化(rarefaction)と嚢胞;矢印は異常白質内の嚢胞性希薄化を示す
B. エカルディ・グティエール症候群のCTにて可視化できるカルシウム沈着とヘモジデリン沈着;MRIで容易には鑑別できない;矢印は基底核の石灰化を示す
C. X連鎖性副腎白質ジストロフィーの異常白質内にみられるT1強調画像における造影効果;矢印は異常白質内の造影効果を示す
D. POLG関連疾患の皮質灰白質病変;矢印は皮質外套の腫大所見と皮髄境界の喪失を示す
E. L-2-ヒドロキシグルタル酸尿症の歯状核にみられる小脳異常;矢印は対称性の高信号を示す
F. 遺伝性痙性対麻痺11の脳梁(特に膝部)の菲薄化;矢印はこの疾患で典型的にみられる前方の嘴様変化を伴う菲薄化した脳梁を示す
G. アレキサンダー病にみられる非石灰化基底核病変;矢印は基底核の高信号を示す
H. 常染色体優性成人発症白質ジストロフィー(ADLD)の典型的な脳幹病変;矢印は脳幹の高信号病変を示す
I. LBSLの脊髄病変;矢印は脊髄内の高信号病変を示すが、可視範囲の脊髄伝導路全長を障害している
特異的検体検査
脳MRIの所見が特定の白質ジストロフィーに一致する場合は,その疾患に対する生化学的あるいは分子遺伝学的検査を検討する(表1).注:分子遺伝学的検査は評価中に得られた情報に基づいて段階的に1つの遺伝子検査を行うか,もし可能であればマルチ遺伝子パネルを用いて行う.
表1
厳密な診断基準を満たす白質ジストロフィー
| 疾患名 | 遺伝形式 | 遺伝子1 | 生化学検査/その他 |
|---|---|---|---|
| 18q欠失症候群 | ほとんどがde novo欠失;まれに遺伝性 | MBPを含む18q微細欠失に対する染色体検査 | |
| 成人ポリグルコサン小体病(APBD) | AR | GBE1 | 筋,神経,腋窩皮膚の組織病理検査:病的なポリグルコサミン蓄積 |
| エカルディ・グティエール症候群(AGS) | 通常AR; まれにAD |
TREX1 RNASEH2A RNASEH2B RNASEH2C SAMHD1 ADAR |
髄液検査:リンパ球増多,インターフェロンα↑,プテリン↑ |
| アレキサンダー病 | AD | GFAP | |
| 常染色体優性成人発症白質ジストロフィー(ADLD) | AD | LMNB1 | |
| 石灰化と嚢胞を伴う脳網膜微小血管症(CRMCC)2 | おそらくAR | 臨床症状および神経放射線学検査 | |
| カナバン病 | AR | ASPA | 尿,血漿,髄液,羊水:尿中N-アセチルアスパラギン酸↑ 皮膚線維芽細胞:アスパルトアシラーゼ酵素活性の欠損 |
| 脳腱黄色腫症 | AR | CYP27A1 | 血漿と髄液:コレスタノール濃度↑,ケノデオキシコール酸↓ 胆汁,尿,血漿:胆汁アルコールおよび複合糖質濃度↑ 線維芽細胞,肝臓,白血球:ステロール 27-ヒドロキシラーゼ活性↓ |
| 中枢神経系髄鞘形成不全を伴う小児失調症/白質消失病(CACH/VWM) | AR | EIF2B1-5 | |
| 遊離シアル酸蓄積症3 | AR | SLC17A5 | 尿、線維芽細胞、ライソソーム:遊離シアル酸↑ |
| フコシドーシス | AR | FUCA1 | 尿オリゴ糖アッセイ:フコース含有複合糖質↑ 白血球あるいは線維芽細胞:α-フコシダーゼ欠損 |
| 基底核と小脳の萎縮を伴う髄鞘形成不全症(HABC) | おそらくAD | TUBB4A | 臨床症状および神経放射線検査 |
| 髄鞘形成不全症と先天性白内障(HCC) | AR | FAM126A | |
| クラッベ病 | AR | GALC 脚注4参照 |
白血球あるいは線維芽細胞:ガラクトセレブロシダーゼ活性欠損 |
| L-2-ヒドロキシグルタル酸尿症 | AR | L2HGDH | 血漿、尿、髄液:L-2-ヒドロキシグルタル酸濃度(およびリジン)↑ |
| 脳幹および脊髄病変と乳酸上昇を伴う白質脳症(LBSL) | AR | DARS2 | |
| 視床および脳幹病変と乳酸上昇を伴う白質脳症(LTBL) | AR | EARS2 | |
| 皮質下嚢胞をもつ大頭型白質脳症(MLC) | AR | MLC1 HEPACAM (MLC2) |
|
| 異染性白質ジストロフィー(MLD) | AR | ARSA | 白血球、線維芽細胞:アリルスルファターゼA活性↓ 尿:スルファチド類↑ |
| PSAP関連MLD5 | PSAP | 白血球、線維芽細胞:アリルスルファターゼA活性正常 尿:スルファチド類↑ |
|
| マルチプルスルファターゼ欠損症(MSD) | SUMF1 | 他のスルファターゼ類の活性↓ 尿:ムコ多糖類↑、尿スルファチド類↑ |
|
| 軸索スフェロイドをもつ遺伝性びまん性白質脳症(HDLS)6 | AD | CSF1R | |
| 眼歯指異形成症(ODDD) | 通常AD まれにAR |
GJA1 | |
| ペリツェウス・メルツバッハー病(PMD) | XL | PLP1 | |
| ペリツェウス・メルツバッハー様病1(PMLD1) | AR | GJC2 | |
| ペルオキシソーム形成異常症、ゼルウィガー症候群スペクトラム(PBD、ZSS)7 | AR | PEX遺伝子群 | 血漿VLCFA、フィタン酸およびプリスタン酸、血漿および尿中のピペコリン酸および胆汁酸濃度がペルオキシソーム病の病型分類に有用 |
| Pol III関連白質ジストロフィー8 | AR | POLR3A POLR3B |
|
| RNAse T2欠損白質ジストロフィー | AR | RNASET2 | |
| ペルオキシソーム脂肪酸β酸化の単一酵素欠損症9 | AR | 二頭酵素欠損症:HSD17B4 | 血漿VLCFA、フィタン酸およびプリスタン酸、血漿および尿中のピペコリン酸および胆汁酸濃度がペルオキシソーム病の病型分類に有用 |
| ペルオキシソームアシルCoAオキシダーゼ欠損症:ACOX1 | |||
| SCPx欠損症:SCP2 | |||
| シェーグレン・ラルソン症候群 | AR | ALDH3A2 | 尿:ロイコトリエンB4の異常代謝産物 培養皮膚線維芽細胞、白血球:脂肪アルデヒド脱水素酵素(FALDH)あるいは脂肪アルコールの欠損:NAD酸化還元酵素(FAO) |
| SOX10関連疾患 | AD | SOX10 | |
| X連鎖性副腎白質ジストロフィー(X-ALD) | XL | ABCD1 | 血漿VLCFAアッセイ:C26:0、C24:0/C22:0比↑、C26:0/C22:0比↑ |
AD=常染色体優性
AR=常染色体劣性
XL=X連鎖性
VLCFA=極長鎖脂肪酸
アルファベット順に疾患を並べた;GeneReviewsにならって命名した
- 遺伝学的検査はこれらの遺伝子の多くにおいて可能である
- この疾患は現在、保存テロメア維持コンポーネント1をコードするCTC1の病原性バリアントによるコーツ病プラスから独立しているようである.
- サラ病を含む:乳児型シアル酸蓄積病、中間型 GALC活性に重要なSapA-dの活性化蛋白の欠損をもたらすPSAPの欠損
- GALC活性に重要なSapA-dの活性化蛋白の欠損をもたらすPSAPの欠損が報告されている
- PSAPの病原性バリアントはARSA活性に重要な活性化蛋白であるSapB-dの欠損をもたらす
- 神経軸索スフェロイドと色素沈着グリアをもつ成人発症白質ジストロフィーとしても知られている:遺伝性びまん性の色素沈着グリアをもつ色素沈着型正染性白質ジストロフィー(pigmentary type of prthochromatic leukodystrophy: POLD)も含まれる
- 新生児型副腎白質ジストロフィー,乳児型レフサム病が含まれる 歯牙低形成,低ゴナドトロピン性性腺機能低下を伴う
- 髄鞘形成不全症(4H症候群);失調,歯牙低形成を伴う髄鞘形成不全症(ADDH);中枢神経系髄鞘低形成を伴う振戦失調(TACH);歯数不足を伴う白質ジストロフィー(LO);小脳萎縮と脳梁低形成を伴う髄鞘形成不全症(HCAHC)を含む
- D-二頭蛋白(DBP)欠損;ステロールキャリア蛋白-2(SCPx)欠損;ペルオキシソームアシルCoA酸化酵素欠損を含む
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
原因遺伝子が特定されている白質ジストロフィーは常染色体優性遺伝形式,常染色体劣性遺伝形式,X連鎖性劣性遺伝形式にて遺伝しうる;他の遺伝形式を呈する疾患も白質ジストロフィーの原因遺伝子が発見されるにつれて認められる可能性がある.
発端者が白質ジストロフィーに関連した特定の症候群の場合には,その状況に対するカウンセリングの適応がある.
患者家族のリスク:常染色体優性遺伝形式
発端者の両親
- 常染色体優性遺伝形式を示す白質ジストロフィーと診断された患者のほとんどは罹患した親を持つことが多いが,家族歴が陰性の場合もある.
- 家族歴を認めない場合は,親が早世した場合,患者家族において常染色体優性遺伝形式の白質ジストロフィーを判別できない場合,親が遅発性の場合,無症候性の親に浸透率の低い変異アレルが存在する場合,de novoの病原性バリアントである場合が考えられる.
発端者の同胞
- 同胞のリスクは発端者の両親の遺伝的状態による
- 発端者の両親の一方が変異アレルを保有する場合,同胞が変異アレルを受け継いでいるリスクは50%である.
発端者の子
- 常染色体優性白質ジストロフィーの患者がその子供に変異アレルを伝える確率は50%である.
患者家族のリスク:常染色体劣性遺伝形式
発端者の両親
- 両親は絶対保因者であり、それぞれ病原性バリアントを1コピー保有する.
- ヘテロ接合体は無症候である.
発端者の同胞
- 受胎時に,発端者の各同胞は25%の確率で罹患者,50%の確率で無症候性キャリア,25%の確率で非罹患者および非キャリアとなる.
- リスクのある同胞が非罹患者であることが判明すれば,その同胞がキャリアである確率は2/3である.
発端者の子
- 子のすべてが絶対保因者である.
患者家族のリスク:X連鎖性遺伝形式
発端者の両親
- 罹患者の父親が疾患を発症することはなく,病原性変異の保因者であることもない.
- 息子が罹患者で他に血縁男性の罹患者がいる女性は絶対保因者(ヘテロ接合体)である.これらの女性は疾患のごく一部あるいは軽度の症状を呈する可能性がある.注:複数の罹患時をもつが他の血縁者に罹患者がいない女性で,白血球DNAにおいて病原性バリアントが特定できない場合は性腺モザイクである.まれに罹患女性の罹患していない父親が性腺モザイクの場合がある.
- 病原性バリアントを保有する罹患女性の母親はX染色体不活性化が有利に働き,非罹患者あるいは軽度の症状にとどまることが考えられる.
- 罹患者が家系内で一人だけの場合はde novo病原性変異の可能性がある.
発端者の同胞.
同胞に対するリスクは母親がキャリアかどうかによる.
- 発端者の母親が病原性バリアントを保有する場合,それを伝える確率は妊娠ごとに50%である.変異を受け継いだ同胞男性は罹患者となる;変異を受け継いだ同胞女性は保因者となり,通常は罹患しない.
- 発端者が単独症例(家系内で勇逸の発症者の場合など)で母親の白血球DNAに病原性バリアントが特定できない場合は,同胞に対するリスクは低いが,母性性腺モザイクの可能性があるため一般人口頻度よりは高い.
発端者の子.
- 罹患男性の娘は絶対保因者で罹患する可能性も否定できない;息子は罹患しない.
- 罹患女性の子供は病原性バリアントを50%の確率で受け継ぐ.
- X染色体不活性化の可能性があるため,女性の表現型は多様な可能性がある.
発端者の他の家族
他の家族に対するリスクは発端者の両親の遺伝状態よる。
遺伝カウンセリングに関連した問題
DNAバンキングは,将来利用する可能性があることを踏まえて,(通常は白血球から調整した)DNAを貯蔵しておくものである.検査手法や,遺伝子,変異,疾患の解明が将来進展する可能性は十分あり得ることなので,患者のDNAを貯蔵しておくことを検討するべきである.
出生前診断と着床前診断
病原性バリアントが家族内の罹患者で同定された場合は,リスク状態にある妊娠に対する出生前診断と白質ジストロフィーに対する着床前診断が可能である.
リソース
GeneReviewsのスタッフは以下に示すこの疾患の罹患者およびその家族の利益を目的とした疾患関連あるいは包括的支持組織あるいはレジストリを選択してきた.GeneReviewsは他の組織により提供された情報に対する責任は負わない.選別の基準に関する情報については,以下を参照のこと.
- Myelin Disorders Bioregistry Project
Email: myelindisorders@cnmc.org
マネジメント
対症療法
白質ジストロフィーの根底にあるメカニズムは多様だが,多くの症候がこの疾患群において類似している.大多数の症例において,根本的治療は可能ではないが,症状に対するマネジメントによりこれらの複雑な疾患患者の苦痛の軽減やケアの改善を図ることができる.
理想的には,白質ジストロフィー患者のケアにあたってはその経験のある多職種専門家による管理が望ましい.
痙性.薬物は筋緊張を調整するため,および筋への神経シグナルをブロックするために用いられる(化学的脱神経).集中的な理学療法は可動性や機能を向上させるために利用される.
錐体外路症状.ジストニアやジスキネジアは著明な身体障害を引き起こしかねない;薬物治療により著明な機能の改善が得られる可能性がある.
失調.リハビリテーションを行うことは大きなサポートとなりうるが,失調に対する特異的な治療は存在しない.
けいれん.けいれんは末期を除いて典型的な抗てんかん薬で治療するとよく,難治性のことはまれである.
認知発達の遅延/白質脳症.学業あるいは就業にあたって,運動障害に関連する制限を回避することを説明しておくことは重要である.代替コミュニケーションは発語困難の対処として利用できる.精神遅滞や高度の運動障害患者に対する施設入所は必要に応じて検討するべきである.
整形外科的問題.股関節脱臼や側弯などの整形外科的問題の予防と治療に注意を払わないといけない.
栄養.嚥下障害や誤嚥の危険性が高まることに起因する呼吸器の問題は疾患の進行とともにみられる.栄養摂取の減少や栄養不良も生じうる.胃瘻造設を行うかどうかは患者の全身状態,予測される疾患の経過,家族や患者の希望に基づいて決定する.
対症療法
いくつかの白質ジストロフィーにおいては原疾患の症状を予防することが可能である:例えば,X連鎖性副腎白質ジストロフィー,クラッベ病,異染性白質ジストロフィーにおいて,初期段階で造血幹細胞移植(HSCT)あるいは骨髄移植(BMT)の施行が有用となりうる.これらの疾患患者に対しては,HSCTあるいはBMTの適応判断のために専門施設に相談すべきである[Eichler et al 2009].
サーベイランス
標準的なサーベイランスには以下が含まれる:
- 成長や栄養状態の評価のために体重と身長を定期的に測定する
- 整形外科合併症を観察するために理学所見,股関節および脊椎のX線写真を施行する
- 痙攣の再発や徴候に関する定期的な病歴聴取を行う
アレキサンダー病において水頭症の進展を観察するなど,疾患によっては特有のサーベイランスが必要である.
避けるべき薬剤/環境
軽度の頭部外傷や感染により疾患が周期的に悪化することを示唆する症例研究によるエビデンスが存在する白質ジストロフィーがある.これは中枢神経系髄鞘形成不全を伴う小児失調症/白質消失病においてのみ明確に報告されているが,可能であればこれらの誘因を避けるほうがよいと思われる.
リスクのある近親者の評価
遺伝カウンセリングの対象となるリスク状態にある血縁者の検査に関連する問題に関しては遺伝カウンセリングの項を参照のこと.
研究段階の治療
臨床研究に関する情報にアクセスするにはClinicalTrials.govを検索すること.更新履歴
-
Gene Reviews著者: Adeline Vanderver,MD, Davide Tonduti, MD, Raphael Schiffmann, MD, Johanna Schmidt, MPH, MGC, CGC, and Marjo S van der Knaap, MD, PhD.
日本語訳者: 吉田誠克(京都府立医科大学 神経内科)
Gene Reviews 最終更新日: 2014.02.06.日本語訳最終更新日: 2017.03.24(in present)