ホスホリラーゼキナーゼ欠損症(糖原病Ⅸ型)
(Phosphorylase Kinase Deficiency)
[ Synonyms: Glycogen Storage Disease Type IX, GSDIX, PhK Deficiency, Phosphorylase b Kinase Deficiency(同義語:糖原病Ⅸ型、GSDⅨ, PhK欠損症、ホスホリラーゼbキナーゼ欠損症)]
Gene Reviews著者: Jennifer Goldstein, PhD, Stephanie Austin, MS, Proya Kishnani, MD, Deeksha Bali, PhD..
日本語訳者: 和田宏来 (県西総合病院小児科/筑波大学大学院小児科)
Gene Reviews 最終更新日: 2011.5.31.日本語訳最終更新日: 2017.2.24
原文 Phosphorylase Kinase Deficiency
要約
疾患の特徴
糖原病Ⅸ型を起こすホスホリラーゼキナーゼ(Phosphorylase kinase, PhK)欠損症は、グリコーゲン分解を主に調節しているホスホリラーゼbキナーゼ酵素の欠損による。PhK欠損症には肝型PhK欠損症(小児期早期の肝腫大および成長遅滞、しばしば認めるが常に認めるわけではない空腹時のケトーシスおよび低血糖を特徴とする)とかなり稀な筋型PhK欠損症(以下を特徴とする:運動不耐、筋痛、筋痙攣、ミオグロビン尿症、進行性の筋力低下)の2型がある。肝型糖原病の症状や生化学的異常は年齢とともに軽快すると思われる。
診断・検査
PhK酵素は4つのサブユニット(α、β、γ、δ)を有し、サブユニットはそれぞれ4つのコピーから構成される。サブユニットαをコードするPHKA1の病原性変異は、まれなX連鎖性の筋型PhK欠損症を起こす。同じくサブユニットαをコードするPHKA2の病原性変異は、もっともよくみられる病型である肝型PhK欠損症(X連鎖性肝型糖原病)を起こす。サブユニットβをコードするPHKBの病原性変異は、常染色体劣性の肝筋型PhK欠損症を起こす。そしてサブユニットγをコードするPHKG2の病原性変異は、常染色体劣性の肝型PhK欠損症を起こす。診断は臨床所見、赤血球/肝臓/筋組織のPhK活性測定によって行い、分子遺伝学的検査によって確定する。
臨床的マネジメント
症候の治療:
肝型PhK欠損症:低血糖は、複合炭水化物や蛋白質を多くした日中の頻回食によって予防することができる。低血糖もしくはケトーシスが存在する場合、経口摂取できるならポリコース(Polycose®)もしくは果汁を与える、もしくはグルコースを経静脈投与する。肝症状(肝硬変、肝不全、門脈圧亢進症など)は対症的に治療する。
筋型PhK欠損症:身体の状態や機能に応じた理学療法を行う。栄養士(metabolic nutritionist)は活動度に応じた血中グルコース濃度の最適化を行う。
一次症状の予防:
肝型PhK欠損症:低血糖もしくはケトーシスが存在する場合、予防のために複合炭水化物や蛋白質を多くした食事を摂取する。
筋型PhK欠損症:出版された情報はほとんどない。
二次合併症の予防:
肝型PhK欠損症:手術によっては術前にグルコースの静注を行い、つづいて術中・術後にも低血糖を予防するために静注する。全身麻酔では悪性高熱症の予防が必要である。
定期検査:
肝型PhK欠損症:代謝内科医もしくは栄養士による定期的な評価を行う。ストレス時(罹病、激しい活動、急速な成長、二次性徴など)や経口摂取量が減った時だけではなく、血中グルコース濃度や血中ケトンをルーチンにモニターする。18歳未満では、肝臓エコーを12-24ヶ月ごとに施行するべきである。加齢とともに、肝疾患の合併症を評価するために、造影CTもしくは造影MRIを考慮すべきである。
筋型PhK欠損症:代謝内科医、栄養士、理学療法士による定期的な評価を行う。
避けるべき薬物や環境:
肝型PhK欠損症:大量の単糖類はグリコーゲンの肝への蓄積を増やすため避ける。長時間の空腹も避ける。著しい脾腫を認める場合には高度のコンタクトスポーツを避ける。インスリンやインスリン分泌促進剤(スルホニル尿素)のような低血糖を起こす薬剤、アルコール(低血糖の誘因となる可能性がある)も避ける。
筋型PhK欠損症:激しい運動、横紋筋融解症を起こしうるサクシニルコリンやスタチンのような薬剤は避ける。
リスクのある親族の検査:
(家族内で病原性変異が判明している場合)分子遺伝学的検査および(家族内で病原性変異が判明していない場合)代謝内科医による評価により、肝型PhK欠損症のリスクのある同胞に対する早期の診断および治療が可能となる。
妊娠管理:
妊娠中の血糖を正常に保つために食事の管理を行う。
遺伝カウンセリング
PHKA2関連肝型PhK欠損症およびPHKA1関連筋型PhK欠損症はX連鎖性遺伝である。PHKB関連肝筋型PhK欠損症とPHKG2関連肝型PhK欠損症は常染色体劣性遺伝である。
- X連鎖性遺伝:母親がキャリアの場合、それぞれの妊娠で遺伝する確率は50%である。病原性変異を受け継いだ男児は発症する。病原性変異を受け継いだ女児はキャリアとなる。男性患者の子どもが女児の場合は全員病原性変異を受け継ぎ、男児の場合は全員受け継がない。家族内で病原性変異が判明している場合、リスクのある女性親族に対する保因者診断や出生前検査を行うことは可能である。
- 常染色体劣性遺伝:罹患者の同胞は、受胎時には25%の確率で罹患者であり、50%の確率で無症候性キャリアであり、25%の確率で罹患者でもキャリアでもない。家族内で病原性変異が判明している場合、リスクのある親族に対する保因者診断や出生前検査を行うことは可能である。
GeneReviews Scope
| ホスホリラーゼキナーゼ欠損症:含まれる臨床型 |
|---|
|
同義語および、もはや使用されない名称については「命名法」を参照。
診断
臨床診断
糖原病Ⅸ型を起こすホスホリラーゼキナーゼ(Phosphorylase kinase, PhK)欠損症は、グリコーゲン分解を主に調節しているホスホリラーゼbキナーゼ酵素(PhK)の欠損による。PhK酵素は4つのサブユニット(α、β、γ、δ)を有し、それぞれ4つのコピーから構成されるが、この酵素の欠損によりかなり幅がある臨床像を呈する。
このレビューの目的上、ホスホリラーゼキナーゼ(PhK)欠損症を肝型PhK欠損症と筋型PhK欠損症に分類した(図1および表1を参照)。肝型PhK欠損症は病原性変異(PHKB, PHKA2, PHKG2)や遺伝形式にもとづいてさらに3つの亜型に分類される。PHKBの病原性変異は肝筋型PhK欠損症を発症することに留意すべきである。しかし、筋病変の症候は軽度か認めないこともある。よって、この亜型はPHKA2およびPHKG2の病原性変異によって起こる肝型PhK欠損症と臨床的に区別できない可能性がある。
図1 ホスホリラーゼキナーゼサブユニットの発現
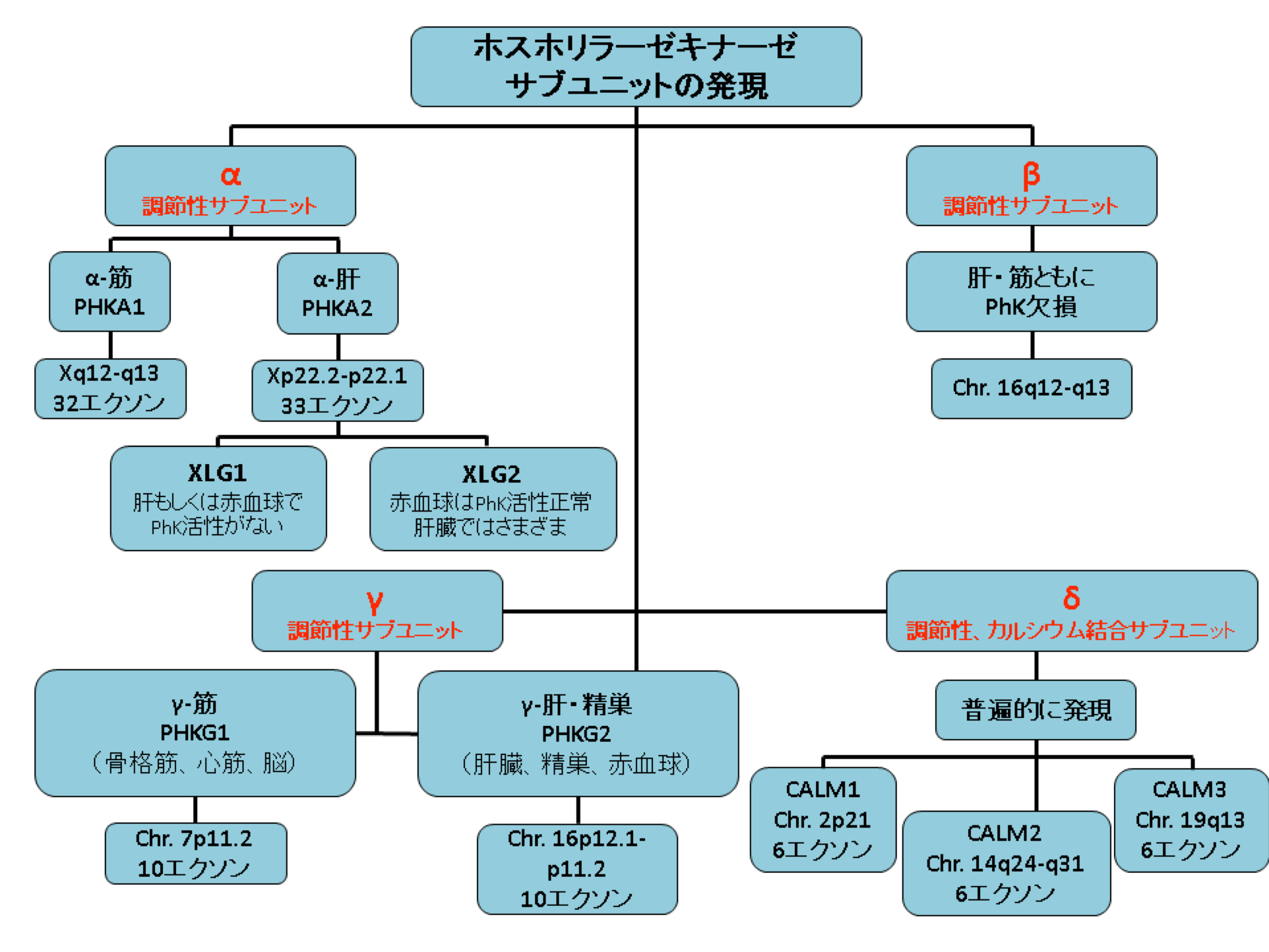
PHKA2関連PhK欠損症はX連鎖性肝型糖原病(X-linked liver glycogenosis, XLG)としても知られ、さまざまな組織の酵素活性にもとづいてXLG1およびXLG2の2つの生化学的亜型に分類される。
表1 ホスホリラーゼキナーゼ(PhK)酵素サブユニットとそれをコードする遺伝子
| 遺伝子 | 全糖原病Ⅸ型における割合 | 罹患組織 | 酵素サブユニット | 遺伝形式 | エクソン数 | 機能 |
|---|---|---|---|---|---|---|
| PhK欠損症に関連する | ||||||
| PHKA1 | ~17%1 筋型PhK欠損症 |
筋 | α | X連鎖性 | 32 | 調節 |
| PHKA2 | ~75% 肝型PhK欠損症 |
肝臓 | α | X連鎖性 | 33 | 調節 |
| PHKB | 不明 | 筋・肝臓2 | β | 常染色体劣性 | 33 | 調節 |
| PHKG2 | 不明 | 肝臓 | γ | 常染色体劣性 | 10 | 触媒 |
| PhK欠損症に関連しないと考えられている | ||||||
| PHKG2 | 不明 | 不明 | γ | 常染色体劣性 | 10 | 触媒 |
| CALM1 | 不明 | 不明 | δ | 常染色体劣性 | 6 | 調節:カルシウム結合 |
| CALM2 | 不明 | 不明 | δ | 常染色体劣性 | 6 | 調節:カルシウム結合 |
| CALM3 | 不明 | 不明 | δ | 常染色体劣性 | 6 | 調節:カルシウム結合 |
- 筋型PhK欠損症の遺伝的基盤は不均一のようである。PHKA1の病原性変異はスクリーニングされた6人のうち1人に認められた。残る患者でPHKA, PHKG1, CALM1, CALM2, CALM3(カルモデュリンをコードする)、PYGM(筋グリコーゲンホスホリラーゼをコードする)、PRKAG2やPRKAG3(肝および筋におけるAMP依存性キナーゼの調節性γ3サブユニットをコードする。AMP依存性キナーゼは直接的もしくは間接的にPhK活性に影響を与える)の病原性変異を認めた者はいなかった。
- PHKB病原性変異による筋型PhK欠損症患者であるにもかかわらず、筋疾患の症候は小児期には存在しないことがあり、主にPHKA2およびPHKG2の病原性変異によって起こる他の肝型PhK欠損症と臨床的に区別できない可能性がある。
肝型PhK欠損症
肝型PhK欠損症には3つの亜型があり、3つの異なる遺伝子(PHKA2, PHKB, PHKG2)の変異によって起こるが、軽度から重度まで幅広いため臨床徴候では区別することはできない。
肝型PhK欠損症は以下のような小児で疑うべきである。
- 肝腫大
- 成長遅滞
- 空腹時ケトーシスおよび低血糖
血液/血清
- 肝トランスアミナーゼは上昇する。
- トリグリセリドおよびコレステロールの軽度上昇を認めることがある。
- 空腹時ケトーシスおよび低血糖、存在する場合は軽度から重度までみられる。
- 尿酸および乳酸値は通常正常だが、一部の患者では上昇しうる。
- 患者のほとんどでグルカゴン反応は正常だが全員ではない。対照群では、グルカゴンは糖新生を促進し、血中グルコース濃度を上昇させる。
肝組織像では、過剰なグリコーゲンの蓄積の結果として肝細胞の膨化が通常認められる。線維性隔壁および低度の炎症性変化も認めることがある。まれだが肝硬変と肝細胞腺腫が報告されている。これまではPHKG2変異患者のみに認められている。しかし、肝硬変はPHKA2のようなPhKをコードする他の遺伝子の病原性変異にも関連している可能性がある。
肝臓グリコーゲン量 肝生検による急速凍結切片の生化学的検査により、構造は正常なグリコーゲンの量が著しく増えているのが認められる。
ホスホリラーゼbキナーゼ(PhK)活性は、ほとんど(全員ではない)の肝型糖原病患者の肝臓、赤血球、白血球において低下している。
- 正常なPhK活性は赤血球では1.0μmol/分/gヘモグロビン、肝臓では0.1μmol/分/mg蛋白である。
- 異常値は正常レベルの10%未満の場合である。
PHKA2もしくはPHKG2いずれかの病原性変異では筋のPhK活性は正常であり、PHKB変異では低下しうる。
筋型PhK欠損症
PHKA1変異が確認された筋型PhK欠損症患者はわずか6人しか認められていない。小児から成人にわたって以下のいずれかの症状を認める場合には疑うべきである。
- 運動不耐
- 筋痛
- 筋痙攣
- ミオグロビン尿症
- 進行性の筋力低下
血清クレアチンキナーゼ(CK)濃度は正常上限を超えるかもしれない。注:正常範囲は検査機関によって異なる傾向にある。
阻血下前腕運動負荷試験は通常正常であり(この検査の詳細については糖原病Ⅴ型を参照)、グリコーゲン分解は最大運動時(無酸素運動)でも正常なことが示唆される。対照的に、最大より低い強度の運動(サイクル試験)ではグリコーゲン分解が鈍化することが報告されている。このことは、最大よりもやや低い強度の運動時において、筋グリコーゲンホスホリラーゼb(ミオホスホリパーゼ)の活性化にPhKが大きな役割を果たすことを示唆している。最大運動時には、ミオホスホリパーゼbを活性化するのにAMP、イノシン一リン酸、無機リン酸塩のような代謝産物で十分なのかもしれない。
筋電図 は通常正常である。
筋の組織像 では筋細胞膜下に過剰なグリコーゲンの蓄積が認められる。
生化学的に測定された筋グリコーゲン量は、正常な構造を保ったまま常に増加している。
ホスホリラーゼbキナーゼ(PhK)酵素活性 は筋で著明に低下しているが、肝臓、血球、線維芽細胞では正常である。酵素PhKは筋のグリコーゲンホスホリラーゼを活性化するため、筋型PhK欠損症患者においてグリコーゲンホスホリラーゼ(ホスホリラーゼ-a)活性は低下しているかもしれない。
分子遺伝学的検査
遺伝子
ホスホリラーゼキナーゼ(PhK)は4つのサブユニット(α、β、γ、δ)から構成されていて、それはX染色体もしくはさまざまな常染色体のいずれかに局在する8つの独立した遺伝子によってコードされている。これらの8つの遺伝子のうち、4つはその病原性変異がPhK酵素欠損を起こすことが知られている(表1)。
X連鎖性の2つの遺伝子は、
- PHKA1、これはまれな病型のX連鎖性筋型PhK欠損症を起こす。
- PHKA2、これはもっともよくみられる病型の肝型PhK欠損症(X連鎖性肝型糖原病、XLG)を起こす。
常染色体劣性の2つの遺伝子は、 - PHKB、これは肝臓・筋両方のPhK欠損を起こすが、主に肝症状が認められ、筋病変は伴わないこともある。
- PHKG2、これは肝臓のPhK欠損を起こす。
遺伝子座異質性のエビデンス
- PHKG1やPHKBの筋エクソン(エクソン26)のような、PhKサブユニットをコードする他の遺伝子は、筋型PhK欠損症の候補遺伝子と考えられている。しかし、これらの遺伝子において病原性変異は現在まで認められていない。
- カルモデュリンをコードする3つの遺伝子(CALM1, CALM2, CALM3)は広く発現しており、肝もしくは筋型糖原病の候補遺伝子である。現在まで、病原性変異は報告されていない(表1を参照)。
臨床検査
PHKA1およびPHKA2(X連鎖性)
- ゲノムDNAのコード領域および関連するイントロン領域のシークエンス解析 これらの2つの遺伝子のシークエンス解析による変異同定頻度に関するデータはまだない。
- PHKA1 1つの報告によると、筋型糖原病患者6人において病原性変異を認めた者はわずか1人で、このことは他の因子(もう一方の遺伝子の病原性変異、検体の取り扱い状況による偽性のPhK活性低下など)が関与している可能性を示唆している。
- PHKA2 病原性変異の同定頻度は不明だが、男性では100%近いことが予期されている。
- PHKA1 1つの報告によると、筋型糖原病患者6人において病原性変異を認めた者はわずか1人で、このことは他の因子(もう一方の遺伝子の病原性変異、検体の取り扱い状況による偽性のPhK活性低下など)が関与している可能性を示唆している。
注:シークエンス解析に先立って施行したPCRで増幅を認めない場合、男性患者のX染色体でエクソン/全遺伝子欠損を認めることが推定される。欠失・重複解析での確認が必要となるかもしれない。シークエンス解析ではキャリア女性のX染色体におけるエクソン/全遺伝子の欠失(巨大欠失)を同定することはできない。
- 欠失/重複解析
- PHKA1 欠失/重複は報告されていない。よって、変異の頻度は不明である。
- PHKA2 肝型PhK欠損症でエクソンの欠失が報告されている。例えば、Fukaoら(2007)はイントロン19からイントロン26にまたがる欠失を報告している。
PHKBおよびPHKG2(常染色体劣性)
- コード領域および関連するイントロン領域のシークエンス解析
- PHKB シークエンス解析で同定できないエクソンの欠失が報告されているため、変異の同定頻度は100%よりもいくぶん低いかもしれない。
- PHKG2 変異の同定頻度は不明だが100%に近いことが予期される。
- PHKB シークエンス解析で同定できないエクソンの欠失が報告されているため、変異の同定頻度は100%よりもいくぶん低いかもしれない。
欠失/重複解析
- PHKB エクソンの欠失は報告されているが、この方法による同定頻度は不明である。
- PHKG2 欠失/重複は報告されていない。そのため、変異の頻度は不明である。
注:筋に発現しているPhKのγサブユニットをコードするPHKG1の病原性変異はいまだ報告されていない。
表2 ホスホリラーゼキナーゼ欠損症で用いられる分子遺伝学的検査の要約
| 遺伝子1(遺伝形式) | 検査方法 | 同定される変異2 | 検査ごとの病原性変異の割合3 |
|---|---|---|---|
| PHKA1(X連鎖性) | シークエンス解析4 | シークエンス変異 | ~100% |
| 欠失/重複解析5 | 現在までない6 | 不明 | |
| PHKA2(X連鎖性) | シークエンス解析4 | シークエンス変異 | 不明 |
| 欠失/重複解析5 | エクソン/全遺伝子欠失 | 不明 | |
| PHKB(常染色体劣性) | シークエンス解析7 | シークエンス変異 | 不明 |
| 欠失/重複解析5 | エクソン/全遺伝子欠失 | 不明 | |
| PHKG2(常染色体劣性) | シークエンス解析7 | シークエンス変異 | ~100% |
| 欠失/重複解析5 | 現在までない6 | 不明 |
- 染色体座位と蛋白については、表A「遺伝子・データベース」を参照。
- アレル変異に関する情報については「分子遺伝学」の項を参照。
- 示した遺伝子の変異の同定に用いられる検査方法の検出能。
- このX連鎖性の遺伝子において、シークエンス解析は男女ともに小さな遺伝子内欠失・挿入、ミスセンス変異、ナンセンス変異、スプライス部位変異を同定することができる。シークエンス解析に先立って施行したPCRで増幅を認めない場合、男性患者のX染色体でエクソン/全遺伝子欠損を認めることが推定される。欠失・重複解析での確認が必要となるかもしれない。シークエンス解析の結果の解釈について考慮すべき問題はこちらをクリック。
- ゲノムDNAのコーディング領域や隣接するイントロン領域に対するシークエンス解析では容易に同定することのできないエクソン/全遺伝子の欠失/重複を識別する検査である。用いられる可能性のあるさまざまな方法には、この遺伝子/染色体領域を含んだ定量PCR、ロングレンジPCR、MLPA(multiplex ligation-dependent probe amplification)法、染色体マイクロアレイ解析(chromosomal microarray, CMA)などがある。
- ホスホリラーゼキナーゼ欠損症の原因となるPHKA1、PHKG2の欠失/重複は報告されていない。
- シークエンス解析で同定される病原性変異の例としては、小さな遺伝子内欠失・挿入、ミスセンス変異、ナンセンス変異、スプライス部位変異などがある。シークエンス解析の結果の解釈について考慮すべき問題はこちらをクリック。
検査戦略
発端者における確定診断
肝腫大および成長遅滞の小児をみた場合は、空腹時ケトーシスや低血糖がなかろうと肝型PhK欠損症を考慮するべきである(「鑑別診断」を参照)。
- 肝型PhK欠損症が疑われる場合、赤血球のPhK活性測定を行うべきである。肝生検は血液検査で診断に至らない場合まで控えることが推奨されるが、一部の専門家は肝腫大の原因を評価するため肝生検を推奨している。この場合、電顕像、グリコーゲン量の測定、PhK活性の測定、他の肝型糖原病(糖原病Ⅵ型やⅢ型など)で低下する酵素活性の測定を行うべきである。偽陽性や偽陰性が報告されているため、PhK活性の測定結果の解釈は慎重に行うべきである。
- 赤血球/肝臓のPhK活性の低下は分子遺伝学的検査によって確認するべきである。どの遺伝子を検査するかは以下によって左右される。
- 性別 男性はほとんどがX連鎖性肝型PhK欠損症(PHKA2)である傾向にある。女性では常染色体劣性亜型(PHKBもしくはPHKG2)が多い傾向にあるが、偏ったX染色体不活性化(skewed X-chromosome inactivation)の結果によるX連鎖性亜型(PHKA2)のヘテロ接合もありうる。
- 家族歴 家族歴がX連鎖性(PHKA2)か常染色体劣性遺伝(PHKBもしくはPHKG2)に一致する場合。
- 近親婚の存在 常染色体劣性亜型(PHKBもしくはPHKG2)が示唆される。
- 重症度(「遺伝子型と臨床型の関連」を参照)
現時点で知られていることは、
- PHKG2変異は全員ではないが肝線維化や肝硬変のリスクを上昇させるようである。
- PHKB変異は、とくに臨床上および生化学検査で軽症の表現型と関連するようである。
- PHKA2変異に関連する臨床型は軽症から重症まで幅広い。
- 赤血球/肝臓PhK酵素活性が正常な場合、PHKA2の分子遺伝学的検査を考慮する。この遺伝子の病原性変異を有する者のかなりの割合で、in vitroのPhK欠損を呈さない(XLG2亜型)からである。
- PHKB変異を有する者において、in vitroの血球(白血球)PhK酵素活性が正常な場合もある。
注:病原性変異の同定によって肝型PhK欠損症の診断が確定した場合、肝生検は必要ではない。肝生検は赤血球酵素解析や分子遺伝学的検査で診断がつかない場合まで控えるべきである。
運動時に筋痙攣やミログロビン尿症を認める、もしくは進行性の筋力低下や筋萎縮を認める小児をみた場合は、筋型PhK欠損症を考慮するべきである。これらの症候はいくつかの異なる疾患においてもみられる(「鑑別診断」を参照)。
- PHKA1の分子遺伝学的検査によって病原性変異が同定される患者は少数である。しかし、家族歴がX連鎖性遺伝に一致する場合、PHKA1の分子遺伝学的検査を行うことが推奨される。筋のPhK活性が低下しPHKA1シークエンス解析で異常を認めない一部の例では、原因は不明である。
- 単一症例(すなわち筋型PhK欠損症の家族歴のない者)では、PhK欠損症や他の鑑別疾患の精査のため、筋生検で組織や酵素活性の評価を行う。PhKの低下を認めた場合、PHKA1の分子遺伝学的検査を行うべきである。
2つのX連鎖性PhK欠損症のリスクのある親族に対する保因者診断 X連鎖性PhK欠損症はPHKA1もしくはPHKA2の病原性変異によって起こるが、先に家族内における病原性変異を同定する必要がある。
注:(1)女性キャリアはこれらのX連鎖性疾患のヘテロ接合である。PHKA2変異のキャリアは軽度の肝腫大、小児期の低身長、生化学的異常を呈することがあり、また理論的には偏ったX染色体不活性化によって重度となる可能性がある。PHKA1変異の女性キャリアは無症状であることが報告されているが、同じく偏ったX染色体不活性化によって症状を呈することもありうる。(2)女性キャリアの同定には以下のいずれかが必要である。(a)先に家族内における病原性変異を同定する。(b)罹患男性で検査を施行できない場合、可能ならば欠失/重複解析を用いる。(3)キャリアかどうか調べるのに酵素を用いた検査は推奨されない。
2つの常染色体劣性PhK欠損症のリスクのある親族に対する保因者診断 PHKBもしくはPHKG2の病原性変異によって起こるが、先に家族内における病原性変異を同定する必要がある。
注:(1)キャリアはこれらの常染色体劣性遺伝疾患のヘテロ接合で、発症リスクはない。(2)キャリアかどうか調べるのに酵素を用いた検査は推奨されない。
リスク妊娠に対する出生前診断や着床前診断(preimplantation genetic diagnosis, PGD)を行う場合、先に家族内における病原性変異を同定する必要がある。
臨床的特徴
臨床像
肝型PhK欠損症 肝型PhK欠損症に対する理解が深まり、臨床上の重症度にはかなり幅があることが明らかとなった。軽症と考えられてきたが、より重症例も報告されるようになった。「遺伝子型と臨床型の関連」も参照のこと。
典型的には、小児患者は生後1年のうちに肝腫大や成長遅滞を呈する。存在するの場合、ケトン性低血糖は通常軽度であるが、重度の場合や反復する場合もある。脂肪酸の酸化や糖新生が促進されることにより、中等度~高度のケトーシスを呈し、血中グルコース濃度は正常範囲内にとどまることもある。
粗大運動発達の軽度の遅れは小児期早期にしばしば認められる。筋緊張低下や筋力低下は、どの遺伝子が変異しているかに関係なく一部の患者で認められる。
少数患者において認知および言語発達の遅れが報告されている。現時点では、これらの遅れがPhK欠損症に直接関連するのか偶発的なのかは明らかではない。
成長遅滞は小児期に最も顕著に認められるが、その後は成長のキャッチアップが認められる。ほとんどは成人期には正常な身長に達する。
二次性徴は遅れるかもしれない。
肝線維化は起こることがあり、まれではあるが肝硬変に進展する(典型的にはPHKG2変異と関連する)。肝細胞腺腫は非常にまれなようである。
尿細管性アシドーシスも一部の患者で報告されている。
肝型PhK欠損症の女性患者では多嚢胞性卵巣はよく認められる。
肝型PhK欠損症患者において心症状は報告されていない。
ほとんどの肝型PhK欠損症患者で、症状や生化学的異常は年齢とともに改善する。持続的なPhK欠損にも関わらず、ほとんどの成人患者は実質的には無症状である。しかし、高齢者における長期的な効果については大々的には研究されていない。縦断的に患者をフォローした場合、長期的な問題がみられることもありうる。
筋型PhK欠損症は小児期から成人期のいずれにも発症し、運動不耐、筋痙攣、筋痛、ミオグロブリン尿症、進行性の筋力低下といった多岐にわたる症状を呈する。重篤な障害は例外的のようで、進行は年単位のこともある。心臓および肝臓は侵されないようである。認知障害を伴う無症候性ミオパチーの男性例が1例報告されており、PHKA1変異に関連する臨床所見は幅広いことが示唆される。しかし、この男性における認知障害は他に原因による可能性がある。
遺伝子型と臨床型の関連
PHKA1変異は筋型糖原病を起こす。PHKA2変異およびPHKG2変異は肝型糖原病を起こす。PHKB変異は肝筋型糖原病を起こす(筋徴候はさまざまである)。
PHKA1 変異のタイプ、筋PhKの残存活性、発症年齢の間に関連性は認められていない。
PHKA2 症状は軽症から重症までみられるが、遺伝子型と臨床的な重症度の間に関連性は認められていない。同じ病原性変異を有している者でさえ、かなり異なった臨床症状を呈しうる。
遺伝子型と生化学的な臨床型の間の関連性に関して、Hendrickxら(1999)は以下のように述べている。
- PHKA2変異の結果としてαサブユニット蛋白量は減少し(蛋白を不安定にするノンセンスおよびフレームシフト変異もしくはミスセンス変異など)、in vitroのPhK低下が見つかるようになる(XLG1生化学的亜型)。
- PHKA2変異はPhK酵素の活性化を阻害し(酵素の調節部位に影響するミスセンス変異もしくは小さなインフレーム挿入/欠失など)、一部の患者ではin vitroのPhK活性は正常となりうる(XLG2生化学的亜型)。
これらのわずかな変化ではPhKの量は正常であるかもしれない。しかし、その変化は、 - PhKと外来基質・内在性基質の相互作用に特異的に影響を与える。
- in vitroでは発見できない、in vivoの調節に対する効果を示す。
- 細胞のタイプに応じてPhK活性のプロテアーゼ感受性(PhKを活性化する)を高める。
- 活性に特異的に影響するサブユニットアイソフォームの組織に応じた相互作用に影響を与える。
Carrièreら(2008)は、PHKA2のミスセンス変異や小さなインフレーム欠失/挿入が蛋白の2つのドメインに集中していることを示した。
- N末端グルコアミラーゼドメインにおいて、病原性変異(主にXLG2を発症する)は予測されるグリコシド結合部位に集まっており、潜在的なPhKαサブユニットの加水分解活性に対して変異が直接的に影響を及ぼす可能性が示唆される。
- C末端カルシニューリンB様ドメイン(ドメインD)において、病原性変異(主にXLG1を発症する)はPhK触媒サブユニットの調節領域と相互作用すると予測される領域や、この相互作用部位をカバーする領域に集まっている。
XLG1やXLG2といった生化学的亜型の分子遺伝学的基盤を確立するためには、さらなる研究が必要である。注目すべきことに、同じPHKA2変異(p.Arg295His)でもin vitroのPhK活性は正常の場合と低下している場合があり、検体の取り扱いや検査方法のような他の因子も生化学的な臨床型に影響を与えうることを示唆している。
PHKB PHKB変異がとくに軽症の臨床的および生化学的病型と関連するという報告はほとんどない。この遺伝子の変異で遺伝子型/臨床型の関連は報告されていない。
PHKG2 軽症の場合もあるようだが、PHKG2変異は肝線維化や肝硬変のリスクがあるより重症の疾患を起こすようである。PHKG2変異のタイプと重症度の間に関連性はないようである。
透率
他の家族が診断された無症状の男性において、酵素活性の測定により肝型PhK欠損症が見つかっている。しかし、これらの方々が研究の時点で成人(それゆえに所見が消失している)であったかどうか、所見(小児期の肝腫大や低身長など)があったかどうかは明らかではない。出現する症状には多様性がある。たとえば、PHKA2のスプライス部位変異を有する小児患者において、6.8歳の時点で認められたのは低身長のみで、肝腫大や生化学的異常は認められていなかった。多様性や浸透率の完全な解明には、さらなる家族研究が必要である。
命名
肝型PhK欠損症 歴史的には、肝型PhK欠損症の分類は糖原病Ⅵa型・Ⅵb型からⅧ型に、そしてⅨ型へと推移した。
注:(1)糖原病Ⅴ型(筋特異的)もしくはⅥ型(肝特異的)を起こすグリコーゲンホスホリラーゼ欠損は、糖原病Ⅸ型を起こすPhK欠損とは異なる。しかし、以前の糖原病の再分類では混乱が生じたかもしれない。PhKはグリコーゲンホスホリラーゼを活性化するため、PhK欠損症ではホスホリラーゼ欠損も起こしうる。(2)糖原病Ⅷ型という分類はもはや存在しない。過去には、糖原病Ⅷ型はPhK欠損症の一部の症例で用いられていた。
肝型PhK欠損症はさらに以下のように分類された。
- 糖原病Ⅸa型、現在ではPHKA2関連糖原病Ⅸ型として知られる。
- 糖原病Ⅸb型、現在ではPHKB関連糖原病Ⅸ型として知られる。
- 糖原病Ⅸc型、現在ではPHKG2関連糖原病Ⅸ型として知られる。
筋型PhK欠損症は糖原病Ⅴb型および糖原病Ⅸd型と呼ばれた。
頻度
肝型PhK欠損症は糖原病全体の約25%を占めると思われ、推定の発生率は100,000人に1人である。しかし、病像は多彩で診断の確定には困難を伴うため、見逃されている可能性がある。
筋型PhK欠損症はまれなようであるが、筋症状が軽度のため見逃されている可能性がある。
PhK欠損症の発生率の高い民族は知られていない。
遺伝学的関連(アレル)疾患
PHKA1, PHKA2, PHKB, PHKG2の変異と関連がある他の臨床型は知られていない。
鑑別診断
以下の疾患は肝型PhK欠損症によく認められる特徴を有する。
糖原病Ⅵ型(GSDⅥ)は肝グリコーゲンホスホリラーゼの欠損によって起こるが、その酵素は肝ホスホリラーゼbキナーゼ(PhK)によって活性化される。糖原病Ⅵ型と肝型PhK欠損症を臨床所見単独で区別することは不可能である。さらに、肝型PhK欠損症患者でもin vitroの肝グリコーゲンホスホリラーゼ活性は低いことがある。この2つの疾患は、酵素検査や分子遺伝学的検査で区別することができる。
糖原病Ⅰ型(GSDⅠ)は成長遅滞、肝腫大、低血糖を特徴とし、通常ケトーシスは伴わない。低血糖は肝型PhK欠損症よりも重篤な傾向にあるが、ときには肝型PhK欠損症でも重篤な低血糖が起こることがある。糖原病Ⅰ型の特徴である血清乳酸値や尿酸値の上昇は、通常肝型PhK欠損症では認められない。糖原病Ⅰb型患者は好中球減少および好中球機能障害による反復性細菌感染も呈する。遺伝形式は常染色体劣性遺伝である。
糖原病Ⅲ型(GSDⅢ)は肝腫大、低血糖、成長遅滞を呈し、年齢とともに改善する。糖原病Ⅲa型では、筋力低下や血清CKの上昇がしばしば認められる。心筋症も起こることがある。遺伝形式は常染色体劣性遺伝である。
以下の疾患は筋型PhK欠損症によく認められる特徴を有する。
糖原病Ⅴ型(GSDⅤ、マッカードル病)は筋グリコーゲンホスホリラーゼ(ミオホスホリラーゼ)の欠損によって起こるが、その酵素は筋PhKによって活性化される。糖原病Ⅴ型と筋型PhK欠損症は、運動で誘発される筋痙攣、ミオグロビン尿症のエピソード、過剰な筋グリコーゲンといった同じような所見を呈する。発症年齢や臨床所見はとても幅がある。しかし、糖原病Ⅴ型患者で記述されている"セカンドウィンド"現象は筋型PhK欠損症患者では認められない。そのような所見がX連鎖性に遺伝することは、筋型PhK欠損症がPHKA1変異によって起こることを示唆している。糖原病Ⅴ型の遺伝形式は常染色体劣性遺伝である。
糖原病Ⅴ型(GSDⅦ、垂井病、ホスホフルクトキナーゼ欠損症)は重度の運動不耐を呈し、通常は小児期に発症し、嘔気、嘔吐、重度の筋痛を合併する。激しい運動は重度の筋痙攣やミオグロビン尿症をきたしうる。他の特徴には、代償性の溶血、高尿酸血症、自発的なセカンドウィンド現象の欠如などがある。遺伝形式は常染色体劣性遺伝である。
筋型PhK欠損症と重複する臨床的特徴を有する他の疾患には以下がある。ミトコンドリアミオパチー(ミトコンドリア異常症概説を参照)、ミオアデニル酸デアミナーゼ欠損症、カルニチンパルミトイルトランスフェラーゼⅡ欠損症、ホスホグリセリン酸キナーゼ欠損症、ホスホグリセリン酸ムターゼ欠損症(糖原病Ⅹ型)、乳酸脱水素酵素欠損症(糖原病?型)、VLCAD欠損症、長鎖3-ヒドロキシアシルCoA脱水素酵素(LCHAD)/ミトコンドリア三機能蛋白欠損症。
致死的な関節拘縮から重度の筋緊張低下までみられるが臓器腫大は伴わない、乳児期発症の重症筋型PhK欠損症も報告されている。これらの患者で分子遺伝学的検査は施行されておらず、その所見がPhK欠損症によるものなのか、それとも二次的なものなのかは明らかではない。
ファンコニ・ビッケル症候群 ファンコニ・ビッケル症候群はGLUT2変異によって起こり、二次的にPhK欠損症を認めることがある。
心臓限局性PhK欠損症 心臓特異的なPhK欠損症はまれな疾患で、生後早期に重篤な限局性の心筋症で発症する。心臓に限局するPhK欠損症の診断には心臓組織の生検が必要である。PhKサブユニットをコードする遺伝子(PHKA1,PHKA2, PHKB, PHKG1, PHKG2, CALM1, CALM2, CALM3-3)の変異はこの臨床型とは関連しないが、一部の患者でPRKAG2(AMP活性化プロテインキナーゼの調節性サブユニットをコードする)変異が認められる。
臨床的マネジメント
初期診断後の評価
肝型PhK欠損症 患者の疾患の広がりとニーズを把握するため、以下のような評価が推奨される。
- 2-3日間の血中グルコース濃度測定(正常は70mg/dLを超える)を、起床時、食前や夜間のコーンスターチ経口摂取の前、活動後に行う。
- 2-3日間の血中ケトン値測定を、起床時、食前や夜間のコーンスターチ経口摂取の前、活動後に行う。血中ケトン値の上昇(β-ヒドロキシ酪酸>1.0mmol/L)は、最適な代謝コントロールや起こりうる低血糖の指標となりうる。
- 過去に行われていなければ、肝臓の画像検査。画像の種類(エコー、MRI、CT)は、年齢や肝臓の状態(肝硬変など)のような因子によって決定する。
- 肝酵素(AST, ALT, ALP)を含む基礎的な代謝検査
- プロトロンビン時間(PT)
- 脂質検査(コレステロール、トリグリセリド)
- 血清CK(肝型PhK欠損症患者の一部では筋症状を認めうる)
- ベースラインの心エコー(PhK欠損症では心臓の問題は報告されていないが[全身の検索があまりされないことや知識不足による]、万一に備えてベースラインの心エコーを施行することがある)
筋型PhK欠損症 患者の疾患の広がりとニーズを把握するため、以下のような評価が提唱される。
- 理学療法的評価
- 血清CK測定
病変に対する治療
肝型PhK欠損症
低血糖は、複合炭水化物や蛋白質を多くした日中の頻回食によって予防することができる。
- コーンスターチの用量は臨床症状に応じて6時間ごとに0.6-2.5g/kgとする。
- 蛋白質は腎機能が正常な限り総カロリーの15-25%(年齢に応じて変更する)とすべきである。蛋白質は糖新生によって代替的にグルコースを供給する。
低血糖もしくはケトーシスの徴候が存在する場合、(経口摂取できるなら)ポリコース(Polycose®)もしくは果汁を摂取し、つづいて複合炭水化物や蛋白質の豊富な軽食をとるべきである。正常に戻ったかを確認するため、血中グルコースやケトン濃度を定期的に測定すべきである。もし経口摂取できないなら、経静脈投与を開始すべきである。血糖70mg/dL以上を維持するためには、グルコースの急速静注(50%グルコース, 利用できない場合は10%グルコース)が必要になるかもしれない。急速静注の後につづいて10%グルコースの静注を行うこともある。
肝硬変、肝不全、門脈圧亢進症など肝症状は対症的に治療する。
筋型PhK欠損症
徴候や症状は、糖原病Ⅲ型のような他の筋型糖原病と同じように治療する。
- 全身状態および身体機能に基づいた理学療法的評価・介入を行う。
- 栄養士(metabolic nutritionist)と協力して、運動や活動のレベルに基づき、血中グルコース濃度のモニタリングや最適化を行う。
一次病変に対する予防
肝型PhK欠損症
低血糖 複合炭水化物や蛋白質を多くした頻回食で低血糖を予防する。一部の患者は低血糖予防にそれ以上の治療を必要としない。しかし、重篤で反復する低血糖を伴う一部の患者は、重症度に応じた1日1-4回の非加熱コーンスターチ(用量は0.6-2.5g/kg)の摂取によって予防することができる。一部の患者は就寝前にのみコーンスターチを必要とすることもある。コーンスターチの必要量は年齢とともに少なくなる傾向にある。
筋型PhK欠損症
筋型PhK欠損症患者の一次病変予防に関して、出版された情報はほとんどない。しかし、定期的な中等度の有酸素運動は有益である可能性がある。強い運動は横紋筋融解症や筋痙攣を誘発する可能性があるため避けるべきである。
二次合併症の予防
肝型PhK欠損症 周術期では、絶飲食にしてから可及的すみやかにグルコース静注を行うべきである。術中や術後も低血糖を予防するためグルコース静注を続ける。通常食を摂取できるようになるまでグルコース静注は徐々に減らしていくべきである。突然の中断は低血糖を起こしうる。
CK上昇やミオパチーを認めることがある肝型PhK欠損症患者で全身麻酔が必要な場合は、悪性高熱症に対する予防措置を行うべきである(著者の個人的な経験)。(悪性高熱症の感受性を参照)
小児期の定期予防接種は推奨されているスケジュールで行うべきである。インフルエンザのような低血糖を起こしうる疾患の予防接種を奨めるべきである。
筋型PhK欠損症 ミオパチーを悪化させ顕在化させうる脂質降下薬(スタチンなど)は慎重に用いるべきである。
CK上昇やミオパチーを認める筋型PhK欠損症患者で全身麻酔が必要な場合は、悪性高熱症に対する予防措置を行うべきである。(悪性高熱症の感受性を参照)
小児期の定期予防接種は推奨されているスケジュールで行うべきである。
定期検査
肝型PhK欠損症
- 肝型糖原病に造詣の深い代謝内科医は医学的な問題がないか経過をみるため、栄養士は食事指導やコーンスターチの必要量の調整のために定期的に評価を行う。
- 代謝内医や栄養士による推奨に従い、血中グルコースやケトン値の定期的な測定を行う。血中グルコースやケトン値は、罹病、激しい活動、急速な成長、二次性徴、妊娠のようなストレス時にも測定すべきである。また、経口摂取量が減った時、食事/コーンスターチの量やスケジュールが変わった時も測定するべきである。
注:脂肪酸の酸化や糖新生が促進されることにより、中等度~高度のケトーシスを呈し、血中グルコース濃度が正常なこともありうる。このように代謝コントロールの指標としてケトンを測定し、さらに系統的に精査を進める必要がある。
- 肝臓の画像検査 18歳未満では、肝臓エコーを12-24ヶ月ごとに施行する。年齢とともに、肝疾患の合併症を評価するため、造影CTもしくは造影MRIを考慮する。
- 心エコーのフォローアップ ガイドラインは存在しない。約2年ごと、症状がある場合はより短い間隔でフォローアップする。
筋型PhK欠損症
・肝型糖原病に造詣の深い代謝内科医は医学的な問題がないか経過をみるため、栄養士は食事指導やコーンスターチの必要量の調整のために定期的に評価を行う。
・理学療法士は症状の進行がないかみるため、運動プログラムを指導するため定期的に評価を行う。
避けるべき薬物や環境
肝型PhK欠損症 患者は以下を避けるべきである。
大量の単糖類はグリコーゲンの肝への蓄積を増やすため避ける。
- 長時間の空腹
- 著しい(中等度~高度の)脾腫を認める場合には高度のコンタクトスポーツは避ける。最終決定は臨床的な判断に基づく。
- インスリンやインスリン分泌促進剤(スルホニル尿素)のような低血糖を起こすことが知られている薬剤。しかし、小児肝型PhK欠損症患者において低血糖を助長する薬剤の文献報告はない。
- アルコール摂取は低血糖の誘因となる可能性がある。
成人肝型PhK欠損症患者の低血糖イベントは比較的まれである。しかし、とくに肝障害を認める患者において、潜在的に低血糖を起こしうる薬剤を用いる場合は注意すべきである。
筋型PhK欠損症 患者は以下を避けるべきである。
- 激しい運動
- 横紋筋融解症を起こしうる薬剤(サクシニルコリンなど)
リスクのある親族の検査
(家族内で病原性変異が判明している場合)分子遺伝学的検査および(家族内で病原性変異が判明していない場合)生後1年以内の代謝内科医による評価により、肝型PhK欠損症のリスクのある同胞に対する早期の診断および治療が可能となる。
遺伝カウンセリングとして扱われるリスクのある親族への検査に関する問題は「遺伝カウンセリング」の項を参照のこと。
妊娠管理
理想的には、女性PhK欠損症患者は医療チームの診療を受け、妊娠前に最適な代謝コントロールを維持することがのぞましい。
拮抗ホルモンの分泌促進とそれによる脂肪分解およびケトーシス、ひいては胎児死亡のリスクを避けるため、妊娠期間を通じて正常血糖を保つことがきわめて重要である。妊娠期間中の適切な食事は各個人によって異なる。一部は普段の健康的な食事を続けるだけでいいのに対し、多くは複合炭水化物や蛋白質の豊富な軽食の回数を増やすかコーンスターチを追加・増量する必要がある。正常血糖を保つため、血中グルコースとケトン値も妊娠期間中は定期的に測定すべきである。糖新生によって代替的にグルコースを供給するためには、十分量の蛋白質が必要である。
研究中の治療法
糖原病全体の低血糖を予防する目的で、コーンスターチの改良・修正版であるスーパースターチ(Superstarch)®が研究されている。
さまざまな疾患に関する臨床試験に関する情報はClinicalTrials.govを参照のこと。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
PHKA2関連肝型PhK欠損症およびPHKA1関連筋型PhK欠損症はX連鎖性遺伝である。
PHKB関連肝筋型PhK欠損症とPHKG2関連肝型PhK欠損症は常染色体劣性遺伝である。
患者家族のリスク
発端者の両親
-
男性患者の父親は罹患していない、もしくは変異のキャリアではない。
家族内に2人以上の患者が存在する場合、男性患者の母親は必然的にキャリアである。
- ある男性が家族内で唯一の罹患者(すなわち単一症例)である場合、その母親がキャリアであるか、罹患男性がde novoの病原性変異を有し、母親はキャリアではない可能性がある。PHKA1およびPHKA2のde novo変異の頻度は明らかではない。de novo変異の症例は報告されていない。
注:ある女性において、子どもが2人以上罹患していて他の血縁者は罹患していない場合、また白血球DNAで家族特異的な変異を同定することができない場合、その女性は生殖細胞モザイクを有する。X連鎖性筋型/肝型PhK欠損症における生殖細胞モザイクの可能性/頻度に関するデータはない。
発端者の同胞
- 同胞のリスクは母親がキャリアかどうかによる。
- 母親がキャリアの場合、それぞれの妊娠で遺伝する確率は50%である。病原性変異を受け継いだ男児は発症する。病原性変異を受け継いだ女児はキャリアとなる。PHKA1変異の女性キャリアの有症状例は報告されていないが、理論的には、偏ったX染色体不活性化が存在する場合に症状がみられる可能性はある。PHKA2変異の女性キャリアは罹患していないことも、軽度の肝腫大を呈することも、(まれに)偏ったX染色体不活性化のパターンに応じてより重度の症状を呈することもある(著者の個人的な経験)。
- 罹患男性が単一症例(すなわち家族内で1人だけ発症)の場合やその母親の白血球DNAで病原性変異を同定することができない場合、同胞に対するリスクは低いものの、母親が生殖細胞モザイクを有する可能性があるため一般集団よりは高い。
発端者(男性)の子ども
- 男性患者の子どもが女児の場合は全員病原性変異を受け継ぎ、男児の場合は全員受け継がない。
発端者の他の家族
-
罹患男性の母方の伯母/叔母はキャリアであるリスクがあり、その伯母/叔母の子どもは性別によるがキャリアである、もしくは罹患している可能性がある。
注:分子遺伝学的検査によってde novoの病原性変異を有する家族構成員を見つけることができ、その情報は拡大家族の遺伝的なリスクを評価するのに有用である。
保因者診断
家族内で病原性変異が判明している場合、リスクのある女性親族に対する保因者診断を行うことは可能である。
患者家族のリスク-常染色体劣性遺伝
発端者の両親
- 小児患者の両親は必然的にヘテロ接合である(すなわち1つの変異アレルのキャリアである)。
- ヘテロ接合(キャリア)は無症状である。
発端者の同胞
- 罹患者の同胞は、受胎時には25%の確率で罹患者であり、50%の確率で無症候性キャリアであり、25%の確率で罹患者でもキャリアでもない。
- リスクのある同胞がひとたび罹患していないと判明した場合、キャリアであるリスクは2/3である。
- ヘテロ接合(キャリア)は無症状である。
発端者の子ども
常染色体劣性PhK欠損症患者の子どもは必然的にPHKB変異もしくはPHKG2変異のヘテロ接合(キャリア)である。
他の家族
発端者の両親の同胞がキャリアであるリスクは50%である。
保因者診断
家族内で病原性変異が判明している場合、リスクのある家族構成員に対する保因者診断を行うことは可能である。
遺伝カウンセリングに関連した問題
早期診断・治療を目的としたリスクのある親族の検査についての情報は、「臨床的マネジメント」「リスクのある親族の検査」を参照のこと。
家族計画
- 遺伝学的リスク評価、保因者診断、および出生前診断の可否などについての議論の最適な時期は妊娠前である。
- 罹患者、キャリア、キャリアのリスクがある若年成人に対して遺伝カウンセリング(潜在的な子どもへのリスクや出産方法の選択肢に関する話し合いなど)を行うことは適切である。
DNAバンクは(主に白血球から調整した)DNAを将来利用することを想定して保存しておくものである。検査技術や遺伝子、変異、あるいは疾患に対するわれわれの理解が将来さらに進歩すると考えられるので、DNA保存が考慮される。
出生前診断および着床前診断
家族内の罹患者でひとたび病原性変異が同定された場合、リスク妊娠に対する出生前検査や着床診断は実施可能な選択肢である。
(PhK欠損症のように)知能には影響せずいくつか治療法のある病態に対する出生前診断の要望は多くない。特に早期診断ではなく妊娠中絶を考慮した検査である場合に、医療従事者や家族の間でも出生前検査に関して視点の違いが存在する可能性がある。ほとんどの施設において、出生前診断に関する決定は両親の選択によると考えるが、これらの問題に関して話し合うことがのぞましい。
更新履歴
-
Gene Reviews著者: ennifer Goldstein, PhD, Stephanie Austin, MS, Proya Kishnani, MD, Deeksha Bali, PhD..
日本語訳者: 和田宏来 (県西総合病院小児科/筑波大学大学院小児科)
Gene Reviews 最終更新日: 2011.5.31.日本語訳最終更新日: 2017.2.24(in present)