1p36欠失症候群
(1p36 Deletion Syndrome)
[同義語: 1p36 モノソミー症候群]Gene Reviews著者: Agatino Battaglia, MD
日本語訳者: 山本裕美、升野光雄、山内泰子、黒木良和(川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
Gene Reviews 最終更新日: 2013.6.6 日本語訳最終更新日: 2018.11.18
要約
疾患の特徴
1p36欠失症候群は、まっすぐな眉毛、深い眼球、顔面中部後退、広く低い鼻稜、長い人中、尖った頤(オトガイ)、大きな大泉門、大泉門閉鎖遅延(77%)、小頭短頭(65%)、内眼角贅皮(50%)、および後方に傾いた低位で形態異常の耳介からなる典型的な頭蓋顔面によって特徴づけられる。他の特徴的な所見には、短指/屈指および短い足が含まれる。様々な程度の発達遅滞/知的障害はすべてに認め、筋緊張低下は95%に存在する。痙攣は、罹患者の44%~58%で起こる。他の所見には、脳構造異常(88%)、先天性心疾患(71%)、眼/視力の問題(52%)、聴力障害(47%)、骨格異常(41%)、外性器異常(25%)、腎臓の異常(22%)が含まれる。
診断・検査
1p36欠失症候群の診断は臨床所見から示唆され、1番染色体短腕の最も遠位のバンド(1p36)の欠失の検出により確認される。通常のG分染法による細胞遺伝学的分析、FISH、またはマイクロアレイ染色体検査(CMA)はすべて欠失を検出するために使用することができる。 しかし、いくつかの複雑な欠失は、CMAによってのみ検出されるかもしれない。
臨床的マネジメント
症状の治療:
発語/コミュニケーション、手話言語の使用、運動発達、認知、および社会生活技能に着目したリハビリテーション/教育プログラム;点頭てんかんのためのATCH治療;他の痙攣のタイプには通常の抗てんかん薬(AEDs);摂食困難のための胃瘻チューブを含む特別な摂食技術および/または器具;心筋緻密化障害に対する標準的な薬物療法;目/視力の問題、骨格異常、聴力障害、甲状腺機能低下症、腎臓の異常に対する標準的ケア。
サーベイランス:
必要に応じたリハビリテーション/教育と治療を調整するための体系的なフォローアップは、時間の経過とともに変化する。
遺伝カウンセリング
1p36欠失症候群は、いくつかの遺伝的機構の1つによる1p36染色体領域の欠失によって引き起こされる。1p36欠失症候群を有する罹患者の約52%が新規の1p36端部欠失を有し、約29%は中間部欠失を、約12%は複数の1p36欠失または、1p36重複を伴う1p36欠失を含む複雑な染色体再配列を、そして約7%が1番染色体の派生染色体を有する(1pテロメア領域が別の染色体末端で置換されている)。家族構成員へのリスクは、欠失の起源の機構に依存する。1p36欠失症候群を持つ子どもがいる家族、または両親の1人が1p36を含む染色体再配列の保因者と知られている家族に対して出生前診断は可能である。
診断
臨床診断
1p36欠失症候群の診断は、特徴的な顔貌、筋緊張低下、精神運動遅延、および発語が乏しいまたは発語が全くないことによって示唆され、1番染色体短腕の最も遠位のバンド(1p36)の欠失が検出されることによって確認される。
典型的な顔の特徴
1p36欠失症候群を有する罹患者の顔貌は、時間が経過しても容易に認識可能なままであり、疾患の特徴を表す(図1参照)。顔の特徴には、まっすぐな眉毛、深い眼球、顔面中部後退、広く低い鼻稜、長い人中、尖った頤(オトガイ)が含まれる。他の頭蓋顔面の特徴は、小頭症、短頭、内眼角贅皮、大きな(出生時に3cmより大きい)大泉門、大泉門閉鎖遅延、および後方に傾いた低位で形態異常の耳介である。
図1.
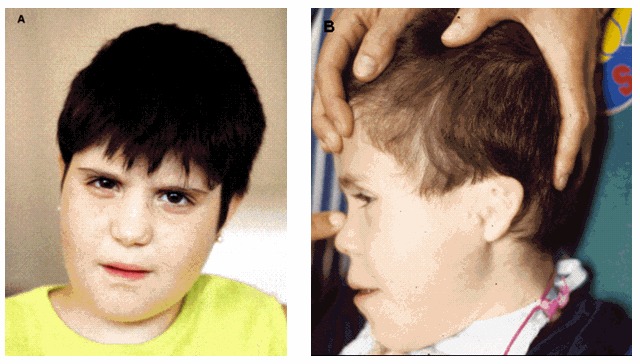
小頭短頭、まっすぐな眉毛、深い眼球、顔面中部後退、広く低い鼻稜、長い人中、尖った頤(オトガイ)、筋緊張低下の顔を認める2人の血縁のない子ども
A. 9.5歳の女児
B. 6.5歳の男児;外耳道閉鎖症と副耳を伴うgrade IIの左側の小耳症も認める
さまざまな程度の発達遅滞/知的障害はすべてに存在する。全身性の筋緊張低下は、罹患者の95%で観察される。
検査
細胞遺伝学的分析:
1p36モノソミーを有する罹患者で同定された再配列の4つのクラスを表1に示す。
- 明らかに「純粋な」端部欠失
- 中間部欠失
- 2つ以上の欠失または重複、三重複、挿入、および/もしくは逆位を伴う欠失を含む、より複雑な再配列
- 不均衡型転座に起因する派生1番染色体
(1)1p36モノソミーを持つ罹患者に共通の切断点または欠失の大きさはない。 (2)細胞遺伝学的に目に見える欠失が真の端部欠失であるか、それともより複雑な再配列であるかを決定するには、特殊な分子細胞遺伝学的手法を用いて達成されるかもしれない(分子遺伝学的検査を参照)。
表1. 1p36欠失症候群に見られる染色体異常
| 染色体異常 | ||||
|---|---|---|---|---|
| 端部欠失 | 中間部欠失 | 複雑な再配列1 | 派生染色体2 | |
| 染色体図 | < |
 |
 |
 |
| 1p36欠失症候群の割合 | 52% | 29% | 12% | 7% |
- 染色体1p36に影響を及ぼす2つ以上の欠失または重複、三重複、挿入、および/もしくは逆位を伴う欠失を含む。
- 1pテロメア領域は、他の染色体末端によって置き換わっている。
分子遺伝学的検査
遺伝子
1p36の発症に関与する領域の遺伝子の欠失は、1p36欠失症候群の唯一の既知の原因である。
臨床検査
表2. 1p36欠失症候群において用いられる細胞遺伝学的および分子遺伝学的検査の概要
| 検査方法1 | 検出されるバリアント | 検査方法によるバリアント検出頻度2 |
|---|---|---|
| 細胞遺伝学的分析3 | 1p36領域の5-Mbより大きい欠失4 | ~25% |
| FISH5 | 1p36領域の100-kbより大きい欠失4 | >95% |
| マイクロアレイ染色体検査による欠失/重複分析6,7 | >95% |
Mb = 106 DNA塩基対;kb = 103 DNA塩基対
- MLPAは、これらの大きさの欠失を検出する推奨方法ではない。
- 示された遺伝子に存在するバリアントを検出するために使用される検査方法の能力。
- 従来のG分染法による細胞遺伝学的分析(通常または高精度)。
- 5Mbより大きい欠失は、5Mbより小さい欠失とほぼ同じ頻度で発生する。
- 少なくとも2箇所のサブテロメア領域の特異的プローブ(Vysis 1p subtelプローブ, Vysis p58 プローブ;D1Z2 Oncor プローブまたはCEB108/T7)を使用したFISHは、親の再配列を同定し、端部および中間部欠失並びに派生染色体を検出し得る。
- ゲノムDNAのコード領域と隣接しているイントロン領域のシークエンス解析では容易に検出できない欠失/重複を同定する検査。使用される手法は以下の通りである。定量的PCR、long-range PCR、多重連結反応依存性プローブ増幅法(MLPA)、およびこの遺伝子断片、あるいは染色体断片を含むマイクロアレイ染色体検査(CMA)。
- 端部欠失、中間部欠失、複雑な再配列および派生染色体は、CMAによって検出される可能性がある。
検査手順
発端者の診断の確定のために。 1p36欠失症候群の疑いのある罹患者を以下のように検査することが適切である:
- 大きな欠失(すなわち>5 Mb)とより複雑な細胞遺伝学的再配列(不均衡型染色体転座)を検出するため
の従来の細胞遺伝学的検討 - 不均衡型転座を検出し、親の染色体再配列を同定するために、少なくとも2箇所のサブテロメア領域の特異的プローブ(Vysis 1p subtelプローブ, Vysis p58 プローブ;D1Z2 Oncor プローブまたはCEB108/T7)を用いたFIS
- より小さな欠失(すなわち<5Mb)もしくは中間部欠失、または複雑な再配列を検出するためのCMAによる欠失/重複分析
(注):
(1)サブテロメアFISHは、使用する2つのプローブが存在するか存在しないを検出する。従って、FISHは(a)プローブの近位の中間部欠失を検出することができない;(b)“真の”端部欠失とより複雑な再配列を区別することはできない;もしくは(c)欠失の範囲を明らかにすることはできない。しかしながら、CMAは3つすべてを検出する可能性がある。(2)MLPAは、欠失/重複分析の一種であり、これらの大きさの欠失を検出するための推奨方法ではない。
リスクのある妊娠に対する出生前診断および着床前遺伝学的診断(PGD)は、発端者における1p36欠失症候群の診断および/または親の均衡状態を事前に確認しておく必要がある。
臨床的特徴
臨床症状
1p36欠失症候群に関連する主要な臨床所見の頻度を表3に要約する。
表3. 1p36欠失症候群における主要臨床所見の頻度
| 所見 | 頻度 |
|---|---|
|
>75% |
|
50%-75% |
|
25%-50% |
|
<25% |
知的障害。発達遅滞と知的障害はこの症候群の特徴である。Battagliaらは罹患者の25%が2~7歳までに開脚歩行で独歩ができることを見出した。約90%が重度から最重度の知的障害を有するが、10%は軽度から中等度の認知障害を有する。75%で表出性言語はなく、残りはいくつかの孤立単語または最初の言語連想のレベルに制限されている。理解は特定の文脈に限られているようである。コミュニケーションの意図は、若年期には限定されているが、ジェスチャーレパートリーの拡張により時間の経過とともに改善する傾向がある。
行動障害は、50%に存在するが、社会的交流不全、かんしゃく発作、手と手首の自咬症、数多くの常同症、そしてそれほど多くはないが過食症が含まれる。
中枢神経系の異常は、罹患者の88%で起こるが、主に側脳室および、くも膜下腔の拡張;皮質萎縮;びまん性脳萎縮;脳梁の低形成、菲薄化、および完全または部分的な欠損を含む。他の報告されている異常には、髄鞘形成の遅延、大脳白質の多巣性高信号域、脳室周囲結節性異所性灰白質、および多小脳回がある。
痙攣は、1p36欠失症候群罹患者の44%~58%で起こる。発症年齢は4日から2年8か月の範囲である。最初の発作は全身性(強直、強直間代、間代性、ミオクロニー)または部分的(単純または複雑)のいずれかである。痙攣を有する全ての人の約20%は、脳波上に認めるヒプスアリスミアと関連する点頭てんかんを有する。点頭てんかんは、現在起こっている発作のタイプであるか、または他のタイプの発作に続いて起こる可能性がある。ほとんどの発作のタイプは、標準的な薬物療法によって良好に制御される。しかし、ある一連の患者では、約1/3の人々が薬物抵抗性てんかんを発症した。
注目すべきは、てんかん性無呼吸は一部の子どもでも起こる可能性がある;サプレッションバーストを伴う早期乳児てんかん性脳症(大田原症候群)もまた罹患者で報告されている。
様々な脳波異常は、ほとんどすべての罹患者に認められる。
摂食困難は、筋緊張低下および/または口腔顔面裂によって引き起こされ得るが、吸啜の困難、嚥下障害とその結果として生じる誤嚥、並びに/または胃食道逆流および嘔吐に関連している。嚥下の研究では、軽度~重度の口腔咽頭嚥下障害が罹患者の72%で観察されている。
先天性心疾患は、罹患者の43%~71%に認められる。報告されている構造上の心臓の異常は、(頻度の順に)心房および心室中隔欠損症、心臓弁異常、動脈管開存症、ファロー四徴症、大動脈縮窄症、右心室の漏斗部狭窄およびエブスタイン奇形である。27%の罹患者に乳児期および小児期の心筋症の病歴を認めた。心筋症は23%が緻密化障害型であり、時間の経過とともに改善する傾向があった。
眼科的異常。斜視、眼振、屈折異常、および視覚無視は、1p36欠失症候群の最も一般的な眼科症状である。白内障、網膜白子症、および視神経欠損が時折観察されている。
1p36欠失症候群を有する罹患者の40%に見られる骨格異常には、骨年齢の遅延、脊柱側弯、肋骨異常、下肢非対称が含まれる。
聴力損失は主に感音性で、1p36欠失症候群の罹患者の47%~82%で認められる。
泌尿生殖器の異常は罹患者の22%に見られ、上極の水腎症を伴う片側腎盂、右腎嚢胞を伴う異所性腎、および片側腎盂拡張が含まれる。
停留精巣、尿道下裂、陰嚢低形成および小陰茎が少数の男性に見られる。
小さな小陰唇、小陰核、大陰唇肥大、子宮発育不全が女性で報告されている。
甲状腺機能低下症は、血中TSHおよびT4レベルが調べられた1p36欠失症候群の様々な年齢の患者の15%~20%で報告されている。
その他。 1p36欠失症候群を有する少数の罹患者で報告された他の異常としては、以下が挙げられる:
- 毛細血管拡張性皮膚病変および色素沈着斑
- 多指
- 先天性脊柱管狭窄
- 先天性筋線維タイプ不均等症ミオパチー
- 後頸部皮膚のたるみ
- 腸回転異常、輪状膵、膵胆管の異常な配置
- 脂肪肝
- 肥厚性幽門狭窄
- 前位肛門または鎖肛、鉤状または二葉胆嚢、小さな脾臓
- 神経芽腫(3人の罹患者)
- 尋常性天疱瘡(1人の罹患者)
遺伝型と表現型の相関
1p36欠失症候群の表現型の変動性を説明するために、研究者らは、1p欠失の大きさと臨床症状の重症度との間の相関を調べた。
Wuら(1999)およびHeilstedtら(2003b)は、ある症状についての発症に関与する領域を同定し、1p36欠失症候群を隣接遺伝子欠失症候群とみなして、完全な遺伝型-表現型の相関を示唆した。しかし、Gajeckaら(2007)は、大規模なコホートにおいて、欠失の大きさと観察された臨床像の数との間に相関がないことを見出した;1p36の小さな(<3Mb)欠失を有する罹患者でさえ、この症候群に関連するよく見られる特徴の大部分を示した。
Redonら(2005)は、1p36欠失症候群に関連する特徴は、隣接遺伝子欠失よりはむしろ、位置効果に起因し得ると仮説を立てた。
有病率
1p36欠失症候群の有病率は、5,000人から10,000人出生あたり1人で、女性対男性比2:1と推定される。
遺伝学的に関連する疾患
このGeneReviewで議論されたもの以外の表現型で、1p36の発症に関与する領域に位置する遺伝子のコピー数の多様性に関連するものは知られていない。
鑑別診断
1p36欠失症候群の臨床的表現型および顔の形態は特徴的である。しかし、以下の疾患と部分的に重なる特徴のために、一部の罹患者が誤診される可能性がある。
- レット症候群は、X連鎖優性遺伝疾患であり、女児は正常な出生と生後6~18か月の間は一見正常な精神運動発達が特徴であり、続いて短期的な発達停滞が起こり、その後、言語および運動能力が急速に退行する。この疾患の特徴は、合目的的な手の機能の喪失と、手の反復常同運動に置き換わることである。自閉的特徴、パニック様発作、歯ぎしり、時折起こる無呼吸および/または過呼吸、失調性歩行および失行、振戦並びに後天性小頭症もまた生じる。この疾患は比較的安定するが、女児が成長するにつれてジストニアや足と手の変形を起こす可能性がある。痙攣はレット症候群の女性の50%で起こる;全身性強直間代発作および複雑部分発作が最も一般的である。突然の原因不明の死亡発生率は上昇する。46,XY核型を有する男性は、2歳前に死亡するような重度新生児脳症となるかもしれない。診断は、古典的症候群について確立された臨床診断基準および/またはMECP2の分子検査に基づいている。
- ・アンジェルマン症候群(AS)は、重度の発達遅滞/知的障害、重度の発語障害、失調性歩行および/または四肢の振戦、並びに頻繁な笑い、笑顔および興奮性を含む不適当な幸福な態度を伴う独特な行動を特徴とする。小頭症および痙攣は一般的である。診断は、臨床的特徴および分子遺伝学的検査および/または細胞遺伝学的分析の組み合わせに基づいている。コンセンサスが得られたASの臨床診断基準が開発されてきた。15q11.2-q13染色体領域における親特異的DNAメチル化インプリンティング解析は、欠失、片親性ダイソミー、またはインプリンティング異常を有するものを含めて、ASを有する罹患者の約78%を検出する;罹患者の1%未満が細胞遺伝学的に目に見える染色体再配列(すなわち転座または逆位)を有する。UBE3Aシークエンス解析は、さらに約11%の罹患者における病的バリアントを検出する。したがって、分子遺伝学的検査(メチル化解析およびUBE3Aシークエンス解析)は、約90%の罹患者における変化を同定する。ASの古典的な表現型特徴を有する罹患者の残りの10%は、現在、同定されていない遺伝学的機構を有しており、従って、診断的検査を施すことができない。
- プラダー・ウィリ症候群(PWS)は、乳児期早期の重度の筋緊張低下および摂食困難が特徴で、その後の乳児期または幼児期の過度の摂食および病的肥満の段階的進展(外的に制御されない限り)へと続く。すべての罹患者は、運動面および言語発達の遅れを伴って、ある程度の認知障害を有する。かんしゃく発作、頑固、融通がきかない、窃盗、偽り、操作的行動、および強迫性性格を伴う特有の行動表現型が一般的である。性腺機能低下症は、男性および女性の両方に存在するが、性器形成不全、不完全な思春期発達、および大部分の症例で不妊として現れる。小さな手足を伴う低身長は一般的である;特徴的な顔貌、斜視および脊柱側弯がしばしば存在し、インスリン非依存性糖尿病はしばしば肥満罹患者において起こる。コンセンサスが得られた臨床診断基準が開発されているが、診断の主流は、15番染色体のプラダー・ウィリ発症に関与する領域(PWCR)内の異常な親特異的インプリンティングを検出するDNAに基づいたメチル化検査である。この検査は、その領域が母親由来だけの遺伝(父親に寄与する領域は存在しない)なのかどうかを決定し、罹患者の99%以上を検出する。メチル化特異的検査は、すべての罹患者にPWSの診断を確認するために重要であるが、特に、非典型的な所見を有するか、または臨床的根拠に基づいて診断を行うのに十分な特徴が現れるには若すぎる患者に重要である。
- スミス・マゲニス症候群(SMS)は、特有の顔貌、発達遅滞、認知障害、および行動異常を特徴とする。顔貌は、幅広く四角い顔、短頭、目立つ前額部、眉毛癒合、軽度眼瞼裂斜上、深い眼球、広い鼻梁、顔面中部後退(以前は顔面中部低形成として知られていた)、短く低い球状の鼻、年齢とともに相対的に下顎前突に変化する乳児期の小顎症、上口唇の反転した唇紅を伴う特有の外観の口である。SMSを有する罹患者は、認知および適応機能において幅広い変動性を有しており、その大多数は、軽度から中等度の知的障害を伴う。行動表現型は、重大な睡眠障害、常同症、並びに不適応および自傷行動を含む。乳児は摂食困難、発育不全、筋緊張低下、反射低下、長時間の昼寝、または食事のために起こされる必要があり、全般的に嗜眠を有する。SMSは、17p11.2の中間部欠失の検出またはRAI1の分子遺伝学的検査のいずれかによって診断される。染色体17p11.2の目に見える中間部欠失は、解像度が適切であれば(550バンド以上)、通常のG分染分析によって共通の欠失を有する全ての罹患者において検出することができる。その変異または欠失がSMSにおける大部分の特徴を説明することが知られている唯一の遺伝子であるRAI1の分子遺伝学的検査は、FISHまたはaCGHによって検出可能な欠失が除外された罹患者に対して行われる。
- アイカルディ症候群(AIS)は、三主徴:脳梁欠損症、特有の網脈絡膜小窩、および点頭てんかんを特徴とする。しかし、現在、いくつかの他の重要な知見が、典型的なアイカルディ症候群をもつ女児に存在することがよく認識されている。神経学的診察では、小頭症、体幹性筋緊張低下、および痙縮を伴う四肢の筋緊張亢進が明らかになり得る。中等度から重度の全般的発達遅滞と知的障害が起こり得る。アイカルディ症候群を有する多くの女児は、生後3か月前からそしてほとんどは1歳前に痙攣を発症する。様々な発作型を伴う進行性の医学的難治性てんかんは、時間の経過とともに発症する。肋骨脊柱異常は一般的であり、罹患者の3分の1までに著しい脊柱側弯をもたらす可能性がある。他の徴候には、特徴的な顔貌、胃腸の問題、小さな手、血管奇形および皮膚の色素病変、腫瘍の発生率の上昇、7~9歳以降の成長率の低下、および思春期早発症または思春期遅発症が含まれる。生存率はかなりばらつきがあり、平均死亡年齢は約8.3歳であり、死亡年齢の中央値は約18.5歳である。
臨床的マネジメント
初期診断後の評価
1p36欠失症候群と診断された罹患者における疾患の程度とニーズを確立するために、以下の評価が推奨される:
- 成長指標の測定と標準成長曲線へのプロット
(注):1p36欠失症候群に特化した成長曲線はない。 - 身体的および神経学的診察
- 認知、言語、および運動発達並びに社会生活技能の評価
- 乳児期における心臓の検査(聴診、心電図、心エコー検査)
- ヒプスアリスミアを伴う点頭てんかんを検出するための覚醒/睡眠時ビデオ・脳波・ポリグラフ検査(主に乳児期)
- 嚥下障害チームへの紹介による栄養の問題と胃食道逆流の評価
- 乳児期もしくは明白な異常がない場合でも診断時の眼科受診
- 骨格異常(例えば、脊柱側弯、下肢非対称)のための身体的診察;異常が見つかれば、整形外科的および理学療法評価のための紹介
- 早期に適切な介入を可能にするために、包括的な耳鼻咽喉科的評価と聴覚スクリーニング(聴性脳幹反応)
- 腎機能検査および構造的な腎臓の異常を検出するための乳児期における腎臓の超音波検査法
- 定期的な甲状腺機能スクリーニング
- 臨床遺伝学の受診
症状の治療
知的障害。運動発達、認知、コミュニケーション、および社会生活技能に注目した個別のリハビリテーションプログラムへの登録が適切である。手話の使用は、コミュニケーション能力を高め、発語の出現を妨げない。早期の介入と、その後の適切な学校の紹介が不可欠である。
痙攣。 1p36欠失症候群を持つ人の25%までが、ヒプスアリスミアを示す脳波に関連する点頭てんかんを発症する。発作はACTHに反応する。
ほとんどの罹患者において、発作タイプはすべて、第一選択薬ができるだけ早く開始されるのであれば、標準的な抗てんかん薬(AEDs)によって良好に制御される。
摂食困難。 口腔運動技能に注意を払った摂食療法が適切である。特別な摂食技術または器具、例えば"Haberman 哺乳瓶"は、口蓋裂のない筋緊張低下の乳児/小児または未修復の口蓋裂を有する乳児/小児の授乳に使用することができる。経管栄養は、嚥下障害のある人に推奨される。胃食道逆流は、標準的な方法で対処されるべきである。ある研究では、1p36欠失症候群を有する少数の罹患者が胃瘻で管理されていた。
先天性心疾患は大抵複雑ではなく、修復しやすい。"緻密化障害"心筋症は標準的な薬物療法に良好に反応する(例えば、フロセミド、カプトプリル、ジゴキシン)。
眼科異常は標準的な方法で治療される。1p36欠失症候群の罹患者の64%までに報告されている視覚無視は、適切なリハビリプログラムで治療することができる。
骨格異常(例えば、脊柱側弯、下肢非対称)は、個別に対処する必要がある。早期治療(理学療法と手術の両方)が推奨される。
聴力損失は、補聴器の試用で治療される。
その他。構造異常(例えば、胃腸、腎臓)は、標準的な方法で対処されるべきである。甲状腺機能低下症は標準的な方法で治療される。
サーベイランス
体系的なフォローアップにより、技能の向上または低下や医療ニーズの変化につれて、リハビリテーションと治療の調整が可能である。
リスクのある血縁者の評価
遺伝カウンセリングの目的でリスクのある血縁者の検査に関連する問題については遺伝カウンセリングを参照。
研究中の治療
種々の疾患に対する臨床試験に関する情報へのアクセスにはClinicalTrials.govで検索。
(注):この疾患には臨床試験がない可能性がある。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝子検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
1p36欠失症候群は、遺伝性または新規の染色体異常の結果であり得る。
患者家族のリスク
発端者の両親
- 1p36欠失症候群の発端者の両親は罹患していないが、1p36を含む均衡型再配列を持つかもしれない(表1参照)。
- 新規欠失の60%が母親由来の染色体に生じる。
- 派生1番染色体を有する罹患者の約3分の1において、派生1番染色体は、親の均衡型転座の分離異常に起因する。
- 1p36欠失症候群の罹患者の両親は、1p36を含む転座を調べる細胞遺伝学的分析を行うべきである。CMAは、再配列が均衡型と予想される場合には検出できないため、これには推奨されない。
- 見かけ上、新規欠失を有する発端者の両親のサブテロメア分析は、片親の1番染色体を含む潜在的な均衡型転座の存在を検出するために適切である。
発端者の同胞
-
発端者の同胞に対するリスクは、両親の遺伝学的状況による。
発端者の欠失が新規の場合、発端者の同胞に対するリスクは、一般集団リスクと同じであるはずである。注目すべきことに、明らかな性腺モザイクが1家系で報告されている。
親が均衡型転座の保因者の場合、1pモノソミー(すなわち、1p36欠失症候群)または1pトリソミーに同胞が罹患するリスクは、一般集団リスクよりも増加する。
発端者の子
1p36欠失症候群の罹患者で子孫を残した人は知られていない。
発端者の他の血縁者
親が染色体再配列の保因者の場合、その家族構成員も染色体再配列の保因者のリスクがある。
保因者検査
発端者の親が均衡型の染色体再配列を有する場合、リスクのある家族構成員は、親の再配列を同定するために用いられる方法(すなわち、染色体分析またはサブテロメアFISH分析)によって検査をすることができる。
遺伝カウンセリングに関連した問題
特定のカウンセリングの問題。
1pおよび他の染色体を含む転座の特定の経験的再発率は不明である。
家族計画
- 遺伝的リスクの決定、出生前検査実施の可能性についての話し合いにおける最適な時期は妊娠前である。
- 染色体再配列の保因者のリスクがあることを知られている若年成人に対しては、(次世代に対する潜在的な
リスク、および生殖の選択肢の議論を含む)遺伝カウンセリングを提供することが適切である。
出生前検査および着床前遺伝学的診断
ハイリスク妊娠。1p36欠失症候群の子どもがいるか、または両親の1人が染色体再配列の保因者であることが知られている家族では、リスクが高い妊娠のための出生前診断および着床前遺伝学的診断が可能である。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- My46 Trait Profile
1p36 deletion syndrome - Chromosome Disorder Outreach (CDO)
PO Box 724
Boca Raton FL 33429-0724
Phone:561-395-4252 (Family Helpline)
Email:info@chromodisorder.org
www.chromodisorder.org - Unique: The Rare Chromosome Disorder Support Group
G1 The Stables
Station Road West
Oxted Surrey RH8 9EE
United Kingdom
Phone: +44 (0) 1883 723356
Email:info@rarechromo.org; rarechromo@aol.com
www.rarechromo.org
分子遺伝学
下記の記述は最新の情報が含まれているため、GeneReviewsに記載されているほかの情報と異なる場合がある
表A.
1p36欠失症候群:遺伝子とデータベース
| 遺伝子 | 染色体座位 | タンパク質 | Clin Var |
|---|---|---|---|
| 該当なし | 1p36 | 該当なし |
データは、以下の標準的な参考文献を編集したものである:HGNCによる遺伝子;OMIMによる染色体座位;UniProtによるタンパク質。リンクを提供したデータベース(Locus Specific, HGMD, Clin Var)の記述については、ここをクリック。
表B.
OMIM に登録されている1p36欠失症候群(OMIMですべてを参照のこと)
| 607872 | CHROMOSOME 1p36 DELETION SYNDROME |
分子遺伝学的病因
1p36欠失症候群を特徴づける臨床像と関連することが確定的に決定された遺伝子はない。
更新履歴
-
Gene Reviews著者: Agatino Battaglia, MD
日本語訳者: 山本裕美、升野光雄、山内泰子、黒木良和(川崎医療福祉大学大学院 医療福祉学研究科 遺伝カウンセリングコース)
Gene Reviews 最終更新日: 2013.6.6 日本語訳最終更新日: 2018.11.18 (in present)
![]()