遺伝性胸部大動脈疾患概説
Heritable Thoracic Aortic Disease Overview)
Gene Reviews著者: Dianna M Milewicz, MD, PhD and Ellen Regalado, MS, CGC.
日本語訳者: 石川亜貴(札幌医科大学医学部遺伝医学)
Gene Reviews 最終更新日: 2017.10.30 日本語訳最終更新日: 2019.2.2
原文: Heritable Thoracic Aortic Disease Overview
要約
目的
遺伝性胸部大動脈瘤と解離(遺伝性胸部大動脈疾患については、このGeneReviewにて簡素化してある)におけるこの概観の目標は、下記の通りである。
目標1 胸部大動脈疾患の臨床像を説明すること。
目標2 遺伝性胸部大動脈疾患の原因と,遺伝子による遺伝性胸部大動脈瘤と解離のリスク評価をレビューすること。
目標3 (可能な場合)発端者の胸部大動脈疾患の遺伝的な原因を特定するための評価の方法を提案すること。
目標4 発端者の家族に遺伝的なリスクの評価を知らせること。
目標5 遺伝的な原因を基にして、胸部大動脈瘤のサーベイランス,および内科的・外科的処置についての管理を(可能な場合)知らせること。
診断的特徴 :遺伝性胸部大動脈疾患
胸部大動脈疾患とは、このGeneReviewにおいては胸部大動脈瘤と大動脈解離(TAAD)を意味する。
遺伝性胸部大動脈疾患(HTAD)は、TAADへ高いリスクを与える遺伝子の変異によって引き起こされる胸部大動脈疾患を意味する(原因参照)。
胸部大動脈瘤は、胸部大動脈の永続的かつ局所的な拡張である。胸部大動脈瘤は、異なる胸部大動脈の部位も巻き込んでいることもある。このレビューでは、大動脈基部や上行大動脈を巻き込む動脈瘤に焦点を当てている(Figure 1参照)。
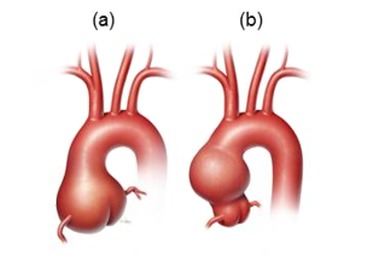
Figure1.
大動脈基部(a)や上行大動脈(b)を巻き込む胸部大動脈瘤
胸部大動脈瘤を評価するために、再構成可能な解剖学的位置で、心エコー、CT、MRIによって大動脈の直径を測定する(血流軸に対して垂直)。経胸壁エコー検査で得られた大動脈の直径の測定値は、CTまたはMRIから得る測定値よりも小さくなる傾向がある[Asch et al 2016]。大動脈基部や上行大動脈の直径を心エコーで評価する検査法は、拡張末期の先端から先端を測定する事になっている[Evangelista et al 2010]。最近のデータは、精密な2次元の経胸壁エコー検査で評価された測定値は(この評価法を用いて)、多裂検出器型CTあるいはMRIにより評価された内径と正確に創刊することを示している[Rodriguez-Palomares et al 2016]。
大動脈基部や上行大動脈について,年齢、性別、体の大きさに基づいた正常大動脈直径のノモグラムが開発されている.[Devereux et al 2012, Kalsch et al 2013]。これらのノモグラムの上限を超える大動脈直径は、拡大または拡張されたとみなされる。
胸部大動脈瘤は、通常は無症候性であり、時間経過とともに拡大する。診断や治療がなされていないと、生命を脅かす急性の上行大動脈解離を引き起こす可能性がある。
大動脈解離は、大動脈の内膜の亀裂によって大動脈の壁の内側や壁に沿って出血を引き起こし、大動脈の中膜が解離する事として定義される。事項の中で、大部分の解離は、大動脈瘤の存在下が起こるが、解離は大動脈の拡大がなくても起こりうる。
大動脈基部は、上行大動脈の関与に基づくStanford分類により区別される。
- A型の解離(Figure 2a, 2b)は、起源部位にかかわらず、上行大動脈を含んでおり、下行胸部大動脈に拡張していても、拡張していなくてもよい。
- B型の解離(Figure 2c)は、典型的にはに左鎖骨下動脈の遠位の下行胸部大動脈で発生し、下行胸部大動脈や腹部大動脈の下のあらゆる距離に広まる。Type Bの解離は、上行大動脈を巻き込まない。
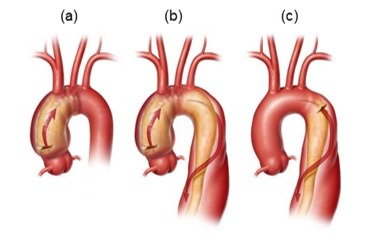
Figure2.
胸部大動脈解離: A型(aとb)と B型(c)
時々大動脈解離を分類するために用いられるDeBakey分類は、解離の起源と、その広がりの程度に基づいて定義される.DeBakey分類についての詳細は、ここをクリック。
自然歴
胸部大動脈瘤は時間とともに大動脈壁が弱くなり、洞上行大動脈移行部で内膜に激しく亀裂が生じるまでは無症状で拡大し、Stanford A型解離を引き起こす。Stanford A型解離では、上行大動脈の内膜壁の裂け目から血流が抜け出し、中膜層(厚い中間)を分離して流れる.
上行大動脈を血流方向(順行性解離)、または基部から逆方向に流れる(逆行性解離)。
過去に、A型大動脈解離では症例の50%以上は突然死を引き起こしたことが報告されているが、最新のデータは、病院死亡率は、特に予防的外科手術や早期の治療を受けている罹患者に減少が見られる[Mody et al 2014]。入院していない罹患者の死亡の大部分は、血液が逆行して心膜嚢を破裂させて心タンポナーデを引き起こす.
急性A型解離(通常、上行大動脈を通って進行し、下行大動脈へ続いて解離を起こした例)を経験している罹患者は、解離した上行大動脈を治療するため、急性または、次に続く緊急手術を伴う高い死亡率がある[Hagan et al 2000, Hiratzka et al 2010]。
B型大動脈解離は、死に至る可能性はあまりなく、また下行大動脈が拡大する可能性がとても少ない傾向にある。
病理学 HTADにおける大動脈病理組織学についての情報は、ここ(pdf)をクリック。
遺伝性胸部大動脈疾患の原因
マルファン症候群、血管型エーラス・ダンロス症候群またはロイス・ディーツ症候群を持たないTAAD罹患者の20%までは、TAADの家族歴がある[Biddinger et al 1997]。
マルファン症候群または他の症候群の臨床診断を受けていないHTADを罹患している家系の約30%は、既知のHTAD関連遺伝子の1つにその原因となる病原性バリアントを有する。
現在までに、16種の遺伝子がTAADに素因となることがわかっている(Table 1)。TAADの家族歴がある多くの家系は、これらの16種の遺伝子の1つに病原性バリアントを持たないため、さらなるHTAD関連遺伝子は、いまだに確認されていない事に留意されたい。
過去に、既知の遺伝的原因がわかっているHTADは、症候性(臨床所見の組み合わせの一部)または非症候性(弧立した所見として起きている)のいずれかであると考えられていたが、遺伝子の病原性バリアントが、症候性から非症候性までの範囲の表現型スペクトルをもたらすことは一般的であるため、HTADの症候性と非症候性の区別がますます曖昧になってきている。
遺伝性胸部大動脈疾患
Table 1.
HTAD関連の遺伝子と表現型
| 遺伝子 (遺伝子座)1 |
この遺伝子2での突然異変を伴うHTADを罹患する家族の割合2 | 症候群 | 確認された他の心室所見 |
|---|---|---|---|
| ACTA2 | 12%-21%3 | Multisystem smooth muscle dysfunction syndrome4 | 注釈4参照 |
| BGN | 稀 | Meester-Loeys syndrome (300989) | |
| COL3A1 | 稀 | エーラス・ダンロス症候群(EDS)Ⅳ型 | |
| FBN1 | 3%5 | マルファン症候群 | |
| FOXE3 | 1.4% 6 | ||
| LOX | 1.5%7 | 大動脈二尖弁、 腹部大動脈瘤、肝動脈瘤 |
|
| MAT2A | 1%8 | 大動脈二尖弁(BAV) | |
| MFAP5 | 0.25%9 | 一部の罹患者に心房細動、僧帽弁逸脱、および動脈蛇行 | |
| MYH11 | 1%10 | 動脈菅開存(PDA)11 | |
| MYLK | 1%12 | ||
| PRKG1 | 1%11 | 一部の患者における冠動脈動脈瘤/解離および動脈蛇行 | |
| TGFB2 | 1%14 | ロイス・ディーツ症候群15 | 腹部大動脈瘤と脳内や他の動脈瘤、解離16,17,18 |
| TGFB3 | 2例の弧発例19、 3世代にわたる1家系20 |
Rienhoff syndrome またはロイス・ディーツ症候群5型 |
|
| TGFBR1 | 3%21 | ロイス・ディーツ症候群15 | |
| TGFBR2 | 5%22 | ロイス・ディーツ症候群15 | 腹部大動脈瘤および/または 頭蓋内および他の動脈瘤および/または解離16,17,18 |
| SMAD3 | 2%23 | 動脈瘤変形性関節症候群; ロイス・ディーツ症候群13 |
|
| [AAT1orFAA1]24 | 不明 | ||
| [AAT2orTAAD1]24 | 不明 |
- 関連する遺伝子が不明の際に遺伝子座は含まれる。
- 頻度は、マルファン症候群、血管型エーラス・ダンロス症候群、ロイス・ディーツ症候群の臨床診断基準に適合しないTAADを罹患する2人以上の家計のコホート調査に基づく。これらの遺伝子に報告されているバリアントの頻度は、集団研究により変化する事に注目する事が重要である。
- Guo et al [2007], Morisaki et al [2009], Disabella et al [2011], Hoffjan et al [2011], Renard et al [2013]
- 早期冠状動脈疾患、虚血性脳卒中、およびモヤモヤ病を伴うTAADは、ACTA2病原体変異体を有する家系において、R258、R118、およびR149残基の変異を有する患者でより頻繁に観察されている[Guo et al 2009 ]。ACTA2 のR179バリアントに起因するmultisystemic smooth muscle dysfunction syndromeの患者には、動脈管塞栓症、動脈硬化症、動脈硬化症、大動脈硬化症、モヤモヤ様脳血管疾患(狭窄および脳血管の拡張)、網膜動脈の蛇行および上腕動脈閉塞がある[Milewicz et al 2010]。
- FBN1[Regalado et al 2015]
- Kuang et al [2016]
- Guo et al[2016]
- Guo et al[2015]
- Barbier et al [2014]
- Pannu et al [2007]
- 動脈菅開存(PDA)は、一定ではない[Glancy et al 2001, Khau Van Kien et al 2004, Khau Van Kien et al 2005, Zhu et al 2006, Pannu et al 2007]。
- Wang et al [2010]
- Guo et al [2013]
- Boileau et al [2012]
- 過去にTGFBR1、TGFBR2、SMAD3、TGFB2の中のヘテロ接合性病原性バリアントに関連する多様な臨床症状は、LDSのⅠ型、Ⅱ型、Ⅲ型を指定しているが、現在、これらの表現型は、臨床的特徴の多様な組み合わせの連続体であることが確認されている。特記として、これらの遺伝子の1つの突然変異を持つ家系において、罹患家系員は、通常、胸部大動脈疾患を有するが、頭蓋内動脈を含む他の血管の蛇行、動脈瘤および解離をも伴う可能性がある。
- TGFBR2とTGFBR1[Nicod et al 1989, Loeys et al 2005, Pannu et al 2005, Loeys et al 2006, Tran-Fadulu et al 2006, Tran-Fadulu et al 2009 ]
- SMAD3[Regalado et al 2011, van de Laar et al 2011]
- TGFB2[Boileau et al 2012, Lindsay et al 2012]
- Rienhoff et al [2013], Matyas et al [2014]
- Bertoli-Avella et al [2015]
- Tran-Fadulu et al [2009]
- Pannu et al [2005]
- Regalado et al [2011]
- AAT1(FAA1)[Vaughan et al 2001]およびAAT2(TAAD1)[Guo et al 2001]として指定された2つの遺伝子座はTAADに関与している。 遺伝子は同定されていない。
ACTA2
胸部大動脈瘤は、一般に紡錘形で、はじめに大動脈基部を巻き込み、その後上行大動脈や大動脈弓に拡張する。
下行大動脈瘤や腹部大動脈瘤は、通常は少ない.胸部大動脈疾患に関連するリスクについては、Table2参照。
ACTA2病原性バリアントのサブセットは、早期発症の脳卒中や冠動脈疾患(男性は55歳未満、女性は60歳未満)、モヤモヤ病のような脳血管疾患にかかりやすい。
アルギニン179残基を破壊するACTA2ミスセンスの病原性バリアントは、平滑筋細胞の機能不全が重度であり,浸透率の高い血管系疾患であり,肺高血圧症、他の臓器での平滑筋細胞の収縮の低下を引き起こす多系統平滑機能不全症候群を引き起こす[Guo et al 2009, Milewicz et al 2010, Munot et al 2012]。全身症状としては、下記のものを含む。
- きな大動脈管開存
- 大動脈縮窄症
- 近位内頸動脈の拡張の特徴的なパターン、脳動脈の閉塞(主に内頸動脈の末端)、頭蓋内動脈の異常な直線的な走行を伴う早期発症の脳血管疾患。脳MRIに観察される脳室周囲の白質高信号領域は、小動脈の閉塞によるものかもしれない[Munot et al 2012]。
- 肺高血圧症
- 四肢の虚血を伴う上腕動脈閉塞[Al-Mohaissen et al 2012]
- 網膜細動脈の蛇行[Moller et al 2012]
- 先天性散瞳
- 腸の蠕動の低下や腸回転異常
- 拡張尿管、腎臓または腎盂、水腎症、膀胱尿菅逆流、再発性の尿路感染症などのさまざまな症状をともなう低緊張性膀胱。プルーンベリーシークエンスも、観察されている [Richer et al 2012, Brodsky et al 2014]。
多形統平滑機能不全症候群と無症候性のHTADを起こすACTA2の分子遺伝学的についての詳細は、ここをクリック(pdf)。
BGN
Meester-Loeys症候群(OMIM 300989)は、大頭症、前頭部突出、眼球突出、眼間開離、眼瞼裂斜下、頬部低形成、皮膚線状を特徴とする。 筋骨格の所見には、頸椎の不安定性、恥骨変形、関節可動域亢進または拘縮、関節脱臼、屈指、、短くてへら状の手指、扁平足を認める.罹患者は、大動脈基部および上行大動脈の動脈瘤および/または解離、および(まれに)肺動脈瘤および/または脳動脈瘤のリスクがある。 脳室拡大が報告されている症例がいくつかある。 ヘミ接合性のBGN病原体バリアントが原因である。 遺伝形式はX連鎖性とされている。
COL3A1
エーラス・ダンロス症候群(EDS)Ⅳ型は、薄く半透明な皮膚、易傷性、特有な顔貌、動脈、腸管、子宮の脆弱性などが特徴である。罹患者は、動脈破裂、動脈瘤、解離のリスクを有するほか、消化管の穿孔あるいは破裂、妊娠中の子宮破裂なども有する。TAADは観察されているが、家系内の1人以上が大~中動脈を巻き込む所見を示す事は非常に稀である。
FBN1
最も一般的なFBN1関連の症候群は、マルファン症候群であり、循環器系の発見により特徴付けられる(バルサルバ洞レベルでの大動脈の拡大、大動脈の亀裂や破壊の傾向(Table 2参照)、逆流を伴うまたは伴わない僧帽弁逸脱、三尖弁逸脱症、近位肺動脈の拡大)。眼科的所見(近視、水晶体偏位)や骨格の所見(体幹に対して異常に長い四肢、関節弛緩症、漏斗胸、鳩胸、脊柱側弯症)。
FBN1病原性バリアントは、眼科的な特徴がなく、マルファン症候群の骨格の特徴や全身の特徴を認めない常染色体優性のTAADを引き起こす可能性もある[Francke et al 1995, Milewicz et al 1996, Stheneur et al 2009, Regalado et al 2016]、それは特にヨーロッパ系の祖先を持つ者と比べて、ヒスパニック系の祖先をもつ者は、骨格的所見がより少ないHTADを有する。[Brautbar et al 2010, Villamizar et al 2010]。
FOXE3
FOXE3のヘテロ接合型ミスセンス変異体が同定された4世代の家系では、TAADの常染色体優性遺伝を有し、女性において多様な臨床表現型および低い浸透性を有していた[Kuang et al 2016]。 罹患者の大部分は、平均年齢45歳(27~63歳)に急性A型解離を認めた。 1人がB型解離を認めた。 病原性であると予測された7つの追加のFOXE3ミスセンス変異体が、家族性TAADを有する個体において同定されたが、臨床的意義をを検証するための家族分析または他の証拠は限られていた。 これらの変異体は、フォークヘッドDNA結合ドメインのC末端にクラスターを形成する。 このドメインのN末端に位置するか、またはこのドメインの外側に位置するほとんどホモ接合病原体変異体は、前節の発生異常および白内障と関連している[Semina et al 2001, Valleix et al 2006, Anjum et al 2010, Doucette et al 2011, Khan et al 2016].
LOX
タンパク質の触媒ドメインおよび、ナンセンス変異体におけるLOXヘテロ接合型ミスセンス変異体は、7家系で報告され、これらの家系においてTAADが共通して認めた。[Guo et al 2016, Lee et al 2016]. 罹患者では、上行大動脈動脈瘤およびA型解離を有していた。 腹部大動脈瘤、肝動脈瘤および二尖大動脈弁が一部で観察されている。 さらに、LOX病原性変異体を有する者には、マルファン症候群の筋骨格所見(例えば、鳩胸、脊柱側弯症、くも状指、高いアーチ型の口蓋)および皮膚線状が様々に存在した。
MFAP5
機能不全のMFAP5病原性変異体は、大動脈基部の拡大と上行胸部大動脈解離を引き起こす可能性がある(Table 2参照)[Barbier et al 2014]。浸透率は低い。
罹患者の中には、心房細動や、僧帽弁逸脱、高口蓋、胸部変形、クモ状指のようなマルファン症候群と重複する臨床的特徴を認める者もいる。
TGFB2、TGFBR1、TGFBR2、SMAD3
胸部大動脈瘤は、一般的に大動脈基部を巻き込む(Table 2参照)。
これらの遺伝子内の病原性変異体は、腹部大動脈、大動脈の動脈分岐部、頭蓋内動脈を含む他の動脈内の動脈瘤や解離に罹りやすくなる。注目すべきことは、著しい大動脈と動脈の蛇行はこれらの遺伝子の変異と関連しているが、大動脈と動脈の蛇行は、HTAD関連の遺伝子の大部分において増加する[Morris et al 2011]。
これらの遺伝子のどれか1つから起きる表現型は、マルファン症候群、ロイス・ディーツ症候群、血管型エーラス・ダンロス症候群の症状を全く認めないものから、最小限認めるものまで範囲がある。
ロイス・ディーツ症候群(LDS)は、早期発症の胸部大動脈疾患、頭蓋内動脈瘤と解離、その他の動脈の動脈瘤と解離、血管蛇行の心血管系の激烈な症状が特徴である。マルファン症候群の全身の特徴(漏斗胸、鳩胸、脊柱側弯症、クモ指症、変形性関節症、関節弛緩症、皮膚線条、ヘルニア)、血管型エーラス・ダンロス症候群の特徴(易傷性、薄く透明な皮膚、萎縮性瘢痕、妊娠中の子宮破裂)、頭蓋顔面の所見を含むLDSに特化する特徴(両眼間開離、二分口蓋垂・口蓋裂、頭蓋縫合早期癒合症、頚椎不安定、内反足、下顎後退症)のさまざまな全身の所見を含む。
TGFB3
Rienhoff症候群あるいは、ロイス・ディーツ症候群5型は、(大動脈基部ないしは上行大動脈の)胸部大動脈瘤か腹部大動脈瘤のいずれかによって、特徴付けられる。A型およびB型解離の両方が観察されている[Rienhoff et al 2013, Bertoli-Avella et al 2015]。大動脈の拡大がほとんど全く認めない解離は報告されていない。他の動脈を含む動脈瘤や解離は稀で、動脈は蛇行しない。大動脈疾患の浸透率は低い。
全ての罹患者ではないが、一部の例においては、骨格的症候(長頭型頭蓋、高アーチ型口蓋、下顎後退症、高身長あるいは低身長、関節過度可動性、クモ状指、胸部変形)、結合組織が関与している証拠所見(僧帽弁逸脱、鼠径ヘルニア、頚椎不安定、内反足奇形)、頭蓋顔面の合併症(両眼間開離、二分口蓋垂、口蓋裂)もみられている。
HTADに併発する細胞経路についての情報はここをクリック。
表現型による遺伝性胸部大動脈疾患
下記の記述は、HTADと関連する遺伝子を持つ人に観察される臨床的特徴のリストである。
- マルファン症候群型の骨格的特徴:BGN, FBN1, LOX, MFAP5, SMAD3, TGFB2, TGFB3, TGFBR1, TGFBR2
- マルファン症候群型の骨格的特徴、水晶体脱臼:FBN1
- マルファン症候群型の骨格的特徴、頭蓋縫合早期癒合症、口蓋裂・二分口蓋垂を伴うロイス・ディーツ症候群:TGFBR1、TGFBR2
- 薄く、半透明な皮膚;易傷性;萎縮性瘢痕:COL3A1、SMAD3、TGFB2、TGFB3、TGFBR1、TGFBR2
- 消化管の破裂;妊娠中の子宮破裂:COL3A1、TGFBR1、TGFBR2
- 動脈管開存(PDA): ACTA2、MYH11、TGFBR2
- 網状皮斑や虹彩異常(iris flocculi):ACTA2
- 先天性瞳孔散大、PDA、大動脈肺動脈窓、モヤモヤ病のような脳血管疾患、肺高血圧症、腸回転異常、低緊張性膀胱:ACTA2、特に179の残基(Arg179)でのアルギニンの変異
遺伝子による胸部大動脈瘤と解離のリスク
Table 2は、16種の既知のHTAD関連の遺伝子のうち、11種と関連する胸部大動脈瘤と解離のリスクに関する情報を表示している。
Table 2.
HTAD: 遺伝子による大動脈疾患のリスク
| 特徴 | 遺伝子 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACTA2 | FBN1 | MAT2A | MFAP5 | MYH11 | MYLK | PRKG1 | TGFB2 | TGFBR1 | TGFBR2 | SMAD3 | ||
| 罹患者の評価 | 数 | n=277 | n=9651 | n=18 | n=10 | n=12 | n=20 | n=37 | n=23 | n=176 | n=265 | n=44 |
| 年齢 | 平均:38歳 (SD 20) |
平均:22歳1 | 中央値:48歳 | 平均:32歳 | 平均:59歳(SD24) | 平均:37歳(SD16) | 平均:42歳(SD17) | |||||
| AD A型 | 頻度 | 71/277 [26%] |
15%1 | なし | 10% | 2/12 [17%] |
7/20 [35%] |
11/37 [30%] |
3/23 [13%] |
31/176 [18%] |
39/265 15%) |
11/44 (25%) |
| 症候が現れる年齢 | 平均:36歳(SD12) | 中央値: 35歳1 | 58歳 | 平均:44歳 (SD6) | 平均:67歳(SD18) | 平均:34歳(SD8) | 平均:45歳(SD13) | 平均:34歳 (SD 17) |
平均: 46歳 (SD 10) |
|||
| AD B型 | 頻度 | 28/277 (10%) |
4%1 | なし | なし | 2/12 (17%) |
2/20 (10%) |
6/37 (16%) |
なし | 4/176 (2%) |
16/265 (6%) |
2/44 (4%) |
| 症候が現れる年齢 | 平均:29歳(SD12) | 中央値:(36歳)1 | 平均:46歳 (SD13) |
18 & 78歳 | 平均:26歳(SD13) | 平均:38歳(SD14) | 平均:46歳(SD10) | |||||
| TAA repair | 頻度 | 16/277 (6%) |
2/18 (11%) |
NR | 1/12(8%) | なし | 5/37 (14%) |
2/23 (9%) |
35/176 (20%) |
63/265 (24%) |
15/44 (34%) | |
| 年齢 | 平均:33歳(SD18) | 平均:44歳 | 22歳 | 平均:43歳(SD12) | 35,60歳 | 平均:33歳(SD16) | 平均:26歳(SD16) | 平均:41歳(SD11) | ||||
| A型 ADが発症した大動脈径 | 中央値:5.75cm (n=24) |
平均:7.2cm (n=158)2 |
NA | NR | 4.4cm (n=1) | 4.0cm (n=1) |
平均:5.0cm (n=2) |
NR | 平均:6.8 cm(n=9) | 平均:5.2 cm(n=13) | 範囲:4.0-6.3cm (n=5) |
|
| ADの累積リスクまたは予防的修復を伴うリスク | 76% (95% CI 64, 86) /85 歳 |
74% (95% CI 67, 81) /60歳 3 |
NR | NR | NR | NR | 55歳で,86% (95% CI 70, 95) | NR | 80歳で 100% |
90歳で 100% 4 |
NR | |
| 大動脈拡張の累積リスク(95%CI) | NR | 60歳で 96% (94, 97)3 | NR | NR | NR | NR | NR | NR | NR | NR | NR | |
| 妊娠関連のADまたは突然死の頻度(妊娠数) | 8/53 女性137人中 | 4/87 女性217人中3 | なし | なし | 0.3(6) | なし | 2/13 女性41人中 | なし | 1/53 女性138人中 | 4/69 女性178人中 4 | なし | |
| リファレンス | Regalado et al[2014] | See footnotes 1-3 |
Guo et al [2015] | Barbier et al [2014] | Pannu et al [2007] | Wang et al [2010] | Guo et al [2013]; Authors, unpublished |
Boileau et al [2012] | Jondeau et al [2016]; Jondeau, personal communication | Jondeau et al [2016]4 ; Jondeau, personal communication | van der Linde et al [2012] | |
AD = 大動脈解離
TAA = 胸部大動脈瘤
NR = 報告なし
NA = 不適応
CI = 信頼区間
- Detaint et al [2010]
- Gott et al [1999]
- Attias et al [2009]
- 大動脈の直径は、主に解離の際の測定を基にしたものである。限定されたデータは、解離後の上行大動脈の直径での増加を示すが、大動脈基部の最小の歪曲を示している.[Rylski et al 2014]。
遺伝性胸部大動脈疾患:不明な原因
HTADの罹患者の約70%は、16種の既知のHTADの遺伝子の1つに病原性変異体を持たない。これらの罹患者は、既知の原因のHTADを罹患する家系員と同様に、大動脈基部または上行大動脈の大動脈瘤や胸部大動脈解離を認めている。
これらの家系についてのその他に観察される所見は以下のものが含まれる。
- 約9%は、頭蓋内動脈瘤、破裂、出血が見られる[Regalado et al 2011]。
- 一部では、大動脈二尖弁(BAV)がTAADと関連する。注目すべきは、家系員には、BAVとTAADを認めるもの、正常な大動脈弁にTAADを認めるもの、またはTAADを伴わないBAVを持つものの可能性がある[Loscalzo et al 2007]。
- 何人かは、PDA、大動脈縮窄、心房中隔欠損症を含む他の動脈閉塞性疾患や先天性心疾患のような他の症候を伴ってTAADが起こるものがいる。[執筆者、個人的な観察]
発端者における遺伝性胸部大動脈疾患の遺伝学的原因を明らかにするための評価策略
HTADの特定の遺伝学的原因を明らかにする事で、リスクのある発端者や血縁者の治療を援助する事ができる(胸部大動脈疾患のリスク評価やサーベイランスと医療介入や外科的処置の推奨)。
TAADの特定の遺伝学的原因を明らかにするには、通常、心臓検査、血管造営の追加、病歴、身体検査、家族歴、分子遺伝学的検査が含まれる。
病歴と身体検査は、TAADの症候性形態に関連する特徴を確認する事に向けられる。
家族歴は、大動脈または他の循環器疾患に注目して、3世代の家族歴を含むべきであり、血縁者の突然の心臓死、分子遺伝学的検査、心血管および身体検査、検死所見、および大動脈病理組織検査の結果を含む診察記録や、直接的な検査結果の書類を含む。
分子遺伝学的検査。HTADの診断は、16種の既知のHTADの遺伝子(Table 1参照)の1つに、ヘテロ接合性の病原性変異体の同定されることで、発端者として確立される。症候群の特徴の症状がみられ、家族歴が陽性、また予期されている年齢よりも若い時に大動脈疾患の発症している、胸部動脈瘤あるいは大動脈解離の罹患した者は、分子遺伝学的検査が根拠となる。
分子遺伝学的検査のアプローチは、標的を絞った遺伝子検査(マルチジーンパネルまたは単一遺伝子検査)とゲノム検査(網羅的ゲノム配列決定)の組み合わせを含む。
標的を絞った遺伝子検査は、臨床医がどの遺伝子が関与している可能性があるかを決定することを必要とするが、ゲノム検査はそうではない可能性がある。症候性や非症候性のTAADの表現型は、重複するので、HTADの大部分の罹患者は、次に示す検査(マルチジーンパネル)、もしくは検査(単一遺伝子検査または包括的ゲノム配列決定)が推奨され、それによって診断される。
推薦される検査
本項で論議されている16種の遺伝子のいくつか、または全てを含むマルチジーンパネルが考慮される。注釈:(1)マルチジーンパネルに含まれる遺伝子、検査方法の感度は、ラボによって差異があるだけでなく、時を経て変化する可能性がある。(2)いくつかのマルチジーンパネルは、本項で論議される状態と関連しない遺伝子を含む。したがって臨床医は、どの多遺伝子パネルが最も合理的なコストで病状の遺伝的原因を特定する可能性が最も高いのかを判断する必要がある。根底にある表現型を説明しない遺伝子における意義が不確定もしくは病原性の変異体の同定を見極める必要がある。(3)いくつかの研究室では、パネルの選択肢には、臨床医が指定した遺伝子を含むカスタマイズされた研究室が設計したパネルおよび/または表現型に焦点を当ててカスタマイズされたエクソーム分析が含まれる。 (4)パネルで使用される方法は、配列分析、欠失/重複分析、および/または他の非シーケンシングに基づく試験を含む。
マルチジーンパネルの紹介はここをクリック。遺伝学的検査を依頼する臨床医のためのより詳細な情報は、ここで見つけることができる。
考慮されるべき検査
単一遺伝子検査。TAADや水晶体偏位を持つ発端者では、FBN1の配列解析に続いて、FBN1を対象とした欠失/重複解析(病原性変異体が配列解析で発見されない場合)を行なうことは、これら2つの所見を有する大多数の患者が、マルファン症候群を有するので妥当である。他の全ての臨床状況で、検討する推薦される検査や他の検査は、症候性HTADの臨床的特徴が広大に重複するため、一般的に単一遺伝子検査の代わりに用いられる事が注意される。
エクソーム解析やゲノム解析を含む包括的ゲノム検査(利用可能な場合)は、表現型のみが遺伝子対象検査が不十分である場合に検討される。包括的ゲノム検査についての詳細は、ここをクリック。
発端者の家族における遺伝子のリスク評価
遺伝性胸部大動脈疾患(HTAD)は、主に常染色体優性の方法で受け継がれる[Milewicz & Regalado 2015]。早急な治療開始と予防対策により恩恵を受ける人をできるだけ早期に特定するために,罹患者であって明らかに無症状であるがリスクある高齢および若年の近親者を評価することは適切である。
HTAD関連遺伝子に既知の病原性バリアントを有する罹患者の家族
分子遺伝学的検査は、両親、同胞,子ども,その他のat riskの遺伝学的状態を明らかにするために推奨される.
- 特定のHTAD遺伝子の病原性バリアントががヘテロ接合性と確認されている人で評価、サーベイランス(監視)、治療を目的とするもの(胸部大動脈疾患の医療管理・外科処置への推薦参照)。
- 特定のHTAD遺伝子の病原性バリアントを持たない人々,したがって、リスクが高くない場合は、病原性バリアントを持つ人に対して示されるようなサーベイランスのレベルから免除される。
HTADの特定の遺伝子の原因が確認されていない罹患者の家族。適切な画像診断は、両親、血縁者、子孫、他のリスクある家族員に提案されるべきである(胸部大動脈疾患の監視参照)。
臨床的マネジメント
この項では、胸部大動脈瘤および解離のリスクアセスメント、胸部大動脈疾患のサーベイランス、および遺伝的原因に基づく(可能な場合)医療および外科的管理のための推奨事項について説明する。
胸部大動脈瘤と解離のリスク評価
HTADの診断時には、関連するリスクを評価するために、標準的な解剖学的位置における大動脈径の測定値を取得するために適切な画像検査を実施する必要がある(Hiratzka et al 2010)(表2参照)。ノモグラムは体表面積(BSA)と性別の正常大動脈径を予測するために確立されています[Devereux et al 2012、Kälschet al 2013]。これらのノモグラムの上限を超える大動脈径は、拡大または拡張と見なされます。
- 心エコー検査は、大動脈基部拡張を検出するために使用されます。再現性のある解剖学的位置で測定を行うべきである:(1)大動脈弁輪、(2)バルサルバ中央洞、(3)洞上行大動脈移行部、および(4)上行大動脈[Hiratzka et al 2010]。
- 心エコー検査がこれらの位置を適切に評価できない場合は、CTまたはMRIを使用して大動脈基部上の拡張を検出できます。測定は以下の場所で行うべきである:バルサルバの大動脈洞、洞上行大動脈移行部(ST junction)、上行大動脈、近位大動脈弓、中大動脈弓、近位下行胸部大動脈(左鎖骨下動脈から約2 cm遠位)、中下行胸部大動脈、横隔膜大動脈、腹腔軸原点の腹部大動脈。
胸部大動脈疾患のサーベイランス
発端者。大動脈拡張の診断後、大動脈の画像法は、大動脈の拡張率を評価するため、6ヶ月後に標準的な解剖学的位置で再計測されるべきである。
- 大動脈の直径が安定している場合、毎年の再検査が実施される。
- 大動脈の直径の変化率が年間0.5cmを超える場合、より頻繁な画像診断を検討されるべきである。
- 上行大動脈や大動脈基部の直径が成人において、約4.0cmを超える場合、より頻繁な再検査が実施される。
検査において罹患者にて同定されているHTAD遺伝子の病原性バリアントが陽性として認められた家族員。サーベイランスは、発端者における胸部大動脈疾患と同様である。
HTADの特定遺伝子の原因が確認されていない罹患者の第一度近親者(例:両親、血縁者、子孫)。発端者の胸部大動脈疾患の種類を最も適切に検出できるイメージングモダリティ(心エコー検査vs CTまたはMRI)を使用して胸部大動脈疾患のサーベイランスを実施する。 例えば、大動脈基部動脈瘤を有する発端者のリスクある近親者は、心エコー検査によってスクリーニングで発見することができる。 対照的に、上行大動脈瘤を有する発端者のリスクある血縁者は、CTまたはMRIでスクリーニングする必要があるかもしれない(心エコー検査が適切に大動脈基部を視覚化できない場合)
胸部大動脈疾患の医学的・外科的管理のための推奨事項
胸部大動脈疾患のガイドラインは公開されている[Hiratzka et al 2010]。
胸部大動脈瘤や解離の治療は、臨床遺伝学者、循環器専門医、心胸郭・脈管外科医を含むHTADに精通している専門家の集学的なチームによる協調的な介入が必要とされる。
胸部大動脈の画像法による早期発見、大動脈径や動脈瘤拡大のサーベイランス、医学的治療、および動脈瘤の適切な時期の予防治療を含む適切な臨床的対応は、胸部大動脈解離に関連する高い罹患率や死亡率を軽減する。
蓄積したデータは、遺伝性胸部大動脈疾患(Table 1)の特定の遺伝子の同定を確認する事を示唆し、いくつかのケースで、病原性バリアントは、胸部大動脈瘤と解離を発症するリスク(Table 2)、予防外科的治療のための大動脈径の最適な範囲(Table 2)、追加の血管疾患のリスクや血管のサーベイランスの必要性などを情報提供できる(Table 1)。
医学的治療
上行大動脈における血行動態のストレスを軽減するために、βアドレナリン遮断薬(例:アテノロール)は、HTADの罹患者に日常的に勧められている[Shores et al 1994, Hiratzka et al 2010]。ロサルタンは、ランダムにロサルタンまたはアテノロールに割り当てられたマルファン症候群の小児および若年成人においてその有効性が示された後、2014年にベータアドレナリン遮断薬の代替薬として追加されている[Lacro et al 2014]。
胸部大動脈疾患の治療ガイドラインでは、一旦、大動脈が拡大したら医療治療を始める事を勧めている。HTAD関連の遺伝子に確認されている病原性バリアントを伴い、大動脈の拡大が見られない個人において、これらの治療を始める事、特にその遺伝子の突然変異が、大動脈の拡大があまり見られない大動脈解離に関連する場合である場合も考慮すべきである(Table 2参照)。
特定の遺伝子の原因が確認されていない場合でも、高血圧は、HTADの罹患者とリスクある家族とともに積極的に治療および管理されるべきである。
喫煙や高脂肪血症を含む他の循環器系のリスク要因についても対処する必要がある。
大動脈の拡大または、HTAD関連の遺伝子に病原性バリアントがある人には運動やコンタクトスポーツを避ける事を勧めるべきである。
大動脈の予防外科的治療
β遮断薬は、胸部大動脈瘤の拡張率を遅らせることができるが、A型胸部大動脈の解離による若年死亡を予防する主軸は、外科的治療である。
一般的に大動脈の直径が正常の約2倍である時には手術が勧められる。この推奨は、大動脈の直径が5.5~6.0cmを超える場合は、待機治療による有害事象(解離、破壊、死亡)が手術を選択するリスクを上回っているという観察に基づいている。しかし、他の研究では、急性A型解離を起こした罹患者の最大60%で、解離時の大動脈の直径が5.5cm未満であったこと、人によっては大動脈の拡張が殆ど見らなかったこと事が指摘されている[Pape et al 2007]。
HTADの特定の原因が確認されている場合 外科的修復の根拠とする大動脈直径に関する胸部大動脈疾患の現在の治療ガイドラインでは遺伝子の特定を推奨する。
遺伝子特定のHTADの最近のデータは、Table 2に要約されている。Table 2のデータは、主に解離した大動脈の測定を基にしている事に留意する事が重要である。解離時に大動脈基部の歪みは最小限であるが、急性解離の結果として上行大動脈がさらに拡大している可能性があることが研究により示されている[Rylski et al 2014]。これらの所見は、遺伝子特異的な外科的管理のためのより良いデータが必要であることを強く示唆する。
以下の4つの遺伝子により遺伝子特定が推奨される。しかし,データは、他の遺伝子において、限定される。
- ACTA2:選択的外科的修復は大動脈根または上行大動脈が最大直径4.5 cmに達したときに考慮すべきです。 [Regalado et al 2015]。
- FBN1:より小さい直径での解離、急速な拡大(すなわち、0.5cm /年を超える)、または有意な大動脈逆流の家族歴がない限り、大動脈根は5cmまでモニターすることができる[Hiratzkaら、2010]。
- TGFBR1またはTGFBR2またはLoeys Dietz症候群:外科的管理は4.0 cmの大動脈基部修復術により積極的である[Williamsら、2007年、MacCarrickら、2014年]。より最近のデータは、そのような積極的な管理がTGFBR1およびTGFBR2を有するすべての患者に必要とされるわけではないことを示している[Tran-Fadulu et al 2009、Jondeau et al 2016]。
大動脈の予防的外科的修復のタイミングにおける他の考察以下のいずれかがある場合に、大動脈径が5.0 cm未満の場合の修復を検討する[Hiratzka et al 2010]。
- 急激な拡大(1年あたり> 0.5 cm)
- 直径5.0 cm未満の大動脈解離の家族歴
- 著しい大動脈弁逆流
HTADの具体的な原因が特定されていない場合、外科的修復は罹患近親者のタイプ大動脈解離時の大動脈径に基づいている(解剖の直前または解剖直前の画像化または死後の所見によって決定される)。
更新履歴
- Gene Reviews著者: Dianna M Milewicz, MD, PhD and Ellen Regalado, MS, CGC.
日本語訳者: 石川亜貴(札幌医科大学医学部遺伝医学)
Gene Reviews 最終更新日: 2017.10.30 日本語訳最終更新日: 2019.2.2 (in present)