SHORT症候群
(脂肪萎縮症を伴うPIK3R1関連インスリン抵抗性症候群)
[SHORT Syndrome]
[Synonym: PIK3R1-Associated Syndromic Insulin Resistance with Lipoatrophy]
Gene Reviews著者: A Micheil Innes, MD, FRCPC, FCCMG and David A Dyment, DPhil, MD, FRCPC, FCCMG.
日本語訳者: 菅原宏美,田村和朗(近畿大学大学院総合理工学研究科 理学専攻 遺伝カウンセラー養成課程)
Gene Reviews 最終更新日: 2014.5.15.日本語訳最終更新日: 2017.8.10.
要約
疾患の特徴
SHORT症候群は、低身長(short stature)、過進展(hyperextensibility)、眼窩の窪み(ocular depression)、Rieger奇形、生歯遅延(teething delay)の頭文字から命名された。SHORT症候群に最も共通してみられる特徴は、軽度の子宮内胎児発育遅延(IUGR)、軽度の低身長、部分的脂肪萎縮症(出生時には顔、後に胸部と上肢。臀部と下肢ではみられないことが多い)、特徴的な顔貌とされている。このほか、しばしばみられる特徴として、Axenfeld-Rieger奇形または関連する前眼房形成不全、生歯の遅れとさまざまな歯科的所見、インスリン抵抗性(典型的には小児期中期から思春期にみられる)と成人初期の糖尿病の両方またはどちらか、感音性難聴がある。現在、分子遺伝学的に診断確定されたのは16家系の患者であり、今後、表現形のスペクトラムや疾患の自然歴の理解が進むことが期待される。
診断・検査
SHORT症候群の診断は、臨床的特徴を持つ発端者で、PIK3R1(PI3Kホロ酵素のサブユニットをコードする)の病原性バリアントがヘテロ接合性に検出された場合に確定する。
臨床的マネジメント
症状の治療:
緑内障に対しては、眼圧を低下させ、安定させることで視力を温存する。
齲歯および歯数の不足については、歯列矯正または義歯を考慮する。
耐糖能障害と糖尿病は、内分泌科医師によるフォローを行う。
感音性難聴に対しては、補聴器を使用する。
サーベイランス:
身長、体重、BMIを含む、成長の定期的なモニタリング。前眼房奇形の有無にかかわらずすべての患者に対し、眼の発生障害や緑内障の管理経験を有する眼科医によって、眼圧測定を含む定期的な眼科検診を行う。インスリン抵抗性と糖尿病のスクリーニングを小児期中期(6-12歳くらい)から開始する。幼児期から定期的な聴力検査を行う。
回避すべき薬剤・環境:
成長ホルモン治療は、インスリン抵抗性を悪化させる可能性がある。
妊娠管理:
インスリン抵抗性/糖尿病の通常の管理を行う。
遺伝カウンセリング
SHORT症候群は、常染色体優性遺伝形式である。新規変異によって発症した患者の割合は明らかではないが、かなりに上ると思われる。SHORT症候群の患児は、50%の確率で病原性バリアントを次の世代に伝える可能性がある。家系内で病原性バリアントが同定されている場合、リスクの高い妊娠に対する出生前診断が可能である。
診断
SHORT症候群の名称は、Gorlin et al [1975]により造られたもので、最初に報告された症例の顕著な特徴のいくつか(低身長、過進展、眼窩の窪み、Rieger奇形、生歯の遅れ)を反映している。しかしながら現在では、これらの5つの特徴は診断に必須ではなく、また必ずしもSHORT症候群に最も特異的な特徴ではないと認識されている。
SHORT症候群で最も共通してみられる症状には、以下のものがある。
- 子宮内胎児発育遅延(IUGR)
- 低身長
- 部分的脂肪萎縮症
- 特徴的な顔の所見。図1を参照。三角形の顔。前額部が突出し、眼窩は窪んでいる。狭い鼻尖と薄い鼻翼。垂れ下がった鼻柱。顔の下半分または下3分の1が相対的に小さい。口角は下向きで顎には窪みがみられることがある。耳は突出しているが耳介低位または後傾はみられない。
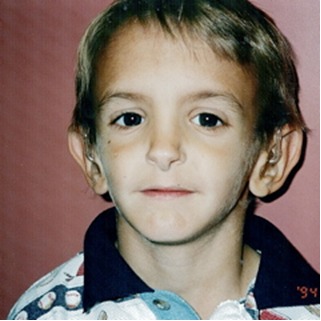
図1 SHORT症候群の顔の特徴
顔の形は三角形で、前額部が突出しており、眼球は深い。鼻は、特徴的な薄い鼻翼と垂れ下がった鼻柱を持つ。口角は下向きで、顎に窪みができることがある。耳は多くは突出しており、典型的な低位ではない。
その他のよくみられる特徴:
Axenfeld-Rieger奇形もしくは関連してみられる前眼房の奇形
- 生歯の遅れ
- インスリン抵抗性/糖尿病
SHORT症候群の公式な診断基準はないが、現在のところ、疾患特異的な顔の形態的特徴により、高い確率でPIK3R1の病的バリアント(ヘテロ接合性)を予測できる。
SHORT症候群の発端者の診断は、臨床的特徴(特に特徴的な顔貌)を持つ発端者に、PIK3R1の病的バリアントがヘテロ接合性に検出された場合に確定する。PIK3R1は、PI3Kホロ酵素のサブユニットの一つをコードする(表1)。
分子遺伝学検査は、PIK3R1の配列解析またはPIK3R1と他の関連遺伝子(「鑑別診断」の項を参照)を含む複数遺伝子パネル検査が中心となる。
注意:複数遺伝子パネル検査に含まれる遺伝子と使用される方法は、検査会社によって、また時期によって異なる。
表1
SHORT症候群の分子遺伝学的検査
| 遺伝子1 | 検査方法 | 発端者での病原性バリアントの検出率 |
|---|---|---|
| PIK3R1 | 配列解析2 | 16家系中163 |
| 欠失/重複解析4 | 報告がない | |
| 未知の遺伝子5 | NA |
NA:該当なし。
- 染色体座位とタンパク質については、表A「遺伝子・データベース」を参照。検出されるアレルバリアントの情報については「分子遺伝学」の項を参照されたい。
- 配列解析では、良性のバリアント、良性と考えられるバリアント、意義不明のバリアント、病原性と考えられるバリアント、病原性のバリアントが検出される。病原性バリアントには、遺伝子内の小さな欠失/挿入、ミスセンスバリアント、スプライス部位バリアントが含まれるが、エクソンや遺伝子全体の欠失/重複は検出できない。配列解析の結果の解釈については、こちらを参照。
- 現在までに、PIK3R1の病原性バリアントはSHORT症候群の16家系で同定されている[Chudasama et al 2013, Dyment et al 2013, Schroeder et al 2013, Thauvin-Robinet et al 2013]。これらの16家系のうち10家系では共通のバリアント(c.1945C>T)が認められている。
- 翻訳領域およびイントロン近傍の配列解析では検出できない、エクソンや遺伝子全体の欠失の検査法。定量的PCR法、long-range PCR法、MLPA法、この遺伝子/染色体を含む領域の染色体マイクロアレイ検査などさまざまな方法がある。
- 現在のところ、PIK3R1は、その病的バリアントがSHORT症候群を引き起こす唯一の原因遺伝子である。特記すべきは、Reardon & Temple [2008]が報告した1人の患者で、SHORT症候群の臨床診断を受けたが、続いて行った検査でPIK3R1の病的バリアントが同定されず、遺伝学的不均質性が示唆されたことである。しかしながら、Axenfeld-Rieger奇形がありSHORT症候群が疑われたもののほかの遺伝子が原因と考えられた症例では、後の検討ではSHORT症候群に特異的な顔の形態的特徴を欠いていたとされる[Dyment et al 2013]。
臨床的特徴
臨床像
現在までに、PIK3R1の病原性バリアントはSHORT症候群の16家系で同定されている[Chudasama et al 2013, Dyment et al 2013, Schroeder et al 2013, Thauvin-Robinet et al 2013]。したがって、現在の(限られた)表現形のスペクトラムや疾患の自然歴についての理解は、今後、報告される患者の増加に従って進んでいくことが期待される。
子宮内胎児発育遅延(IUGR)
SHORT症候群の乳児は、通常、満期または満期の少し前に生まれ、軽度のIUGRを示すことが多い [Lipson et al 1989]。
低身長
幼児期の摂食困難と、十分なカロリー摂取にもかかわらずみられる成長障害のどちらかまたは両方が、よく報告されている。
軽度から中等度の低身長は、小児期を通じてみられる。骨年齢は、遅延することもしないこともある。その他の骨格変化としては、細長い骨幹、大きな円錐形の骨端がある [Haan & Morris 1998]。
軽度の低身長は、これまで報告されたほとんどの成人で認められている。分子遺伝学的にSHORT症候群と診断された成人男性の身長は155cmから163cm、成人女性では143cmから160cmであった [Chudasama et al 2013, Dyment et al 2013, Thauvinet-Robinet et al 2013]。
部分的脂肪萎縮症
部分的脂肪萎縮症は、SHORT症候群に共通してみられる [Koenig et al 2003]。
皮下脂肪の欠失は顔で明らかであり、後に胸部と上肢であらわれやすい。通常、臀部と下肢では起こらないとされるが、肘と臀部で限局性の脂肪萎縮症がみられたとの報告がある[Aarskog et al 1983, Koenig et al 2003]。手でも皮下脂肪の欠失がみられ、皮膚は老化し半透明の外観を呈する。
体型は痩せ型と記述される。これまでの報告では、分子遺伝学的診断が確定した4人の成人男性は全員BMIが18.5未満(範囲:13.5-17.9)、女性は8人中4人が18.5未満(範囲:13.5-22.5)であった [Chudasama et al 2013, Dyment et al 2013, Thauvin-Robinet et al 2013]。
特徴的な顔貌
SHORT症候群に特有の顔の特徴は、時折、老化した、または早老性の顔貌と表現される [Koenig et al 2003]。出生時からみられ、成長とともにより明確になっていく。頭の形は正常で、前後頭囲はほかの成長指標と比例する。
インスリン抵抗性/糖尿病
小児期中期または思春期にインスリン抵抗性が明らかになることがあるが、糖尿病は一般には成人期初期まで発症することはない [Aarskog et al 1983, Schwingshandl et al 1993]。
Axenfeld-Rieger奇形
Axenfeld-Rieger奇形または関連する前眼房奇形または前眼房形成不全は、SHORT症候群患者の多くで報告されてきた。緑内障は、出生時の報告が少なくとも1例あり[Brodsky et al 1996]、後に発症することもある [Bankier et al 1995]。緑内障は、前眼房の房水排出構造が未発達なために起こると考えられている。
PIK3R1の病的バリアントを持つ患者の大部分は、少なくとも近視、遠視、乱視などの眼の問題をいくつか持ち、半数はRieger奇形または関連する前眼房の障害を持つ [Chudasama et al 2013, Dyment et al 2013, Schroeder et al 2013, Thauvin-Robinet et al 2013]。
生歯の遅れ
今までのところ、生歯の遅れは分子遺伝学的に診断されたSHORT症候群患者全員で認められている。これ以外の歯科的所見として、歯数不足、エナメル質低形成、不正咬合がある。複数の齲歯が報告されている [Koenig et al 2003]。
その他
- 患者の約25%で感音性難聴がみられる[Toriello et al 1985, Joo et al 1999, Koenig et al 2003]。80-90dBの難聴は1歳になるまでに診断される。分子遺伝学的に診断が確定したSHORT症候群の患者のうち2人に難聴が認められ、どちらも頻度の高いc.1945C>Tバリアントを保有していた [Dyment et al 2013]。
- SHORT症候群では認知機能への影響は認められないが、軽度の言語遅滞を示す小児患者もいる。
- 全員ではないが一部の患者では、関節の過進展と鼠径部ヘルニアの両方またはどちらかが認められる。
- 生命にかかわる感染症リスクの増加や臨床的に明らかな免疫不全はみられないが、疾患と関係しない頻回の感染歴の報告がある[Koenig et al 2003]。
- 腎石灰沈着が、分子遺伝学的に診断された母親と息子のペアで報告されている[Reardon & Temple 2008]。腎石灰沈着は息子が生後2ヵ月の時、偶然に(直腸肛門閉鎖症のフォローアップ検診での腹部超音波検査によって)発見された。この腎石灰沈着は、2歳時に再評価した際に悪化はみられなかった。母親も、成人してから腎石灰沈着を起こしていた。
- 肺動脈弁狭窄症と異所性腎の報告もある[Koenig et al 2003, Schroeder et al 2013]。
- SHORT症候群では一般的に生殖能力は保たれる。女性のSHORT症候群患者で卵巣嚢胞の報告がある。
遺伝子型と臨床型の関連
これまでのところ、遺伝子型と臨床型の関連は明らかでない。しかしながら、PIK3R1遺伝子の病原性バリアントは、タンパク質C末端側SH2ドメインをコードする領域に集積している。
頻度の高い病原性バリアントc.1945C>Tが、SHORT症候群の発端者16人中10人で同定されている。このバリアントを持つ患者は、通常、典型的なSHORT症候群である一方、診断された患者数が少ないため、ほかの病原性バリアントを持つ患者と表現形に違いがあるかどうかを判断することができない。
浸透率
SHORT症候群は、これまでに分子遺伝学的検査を実施したすべての人々で、完全浸透を示す。孤発例(家族内で1人だけの患者)で両親の検査が可能であったすべてのケースでは、PIK3R1遺伝子の病原性バリアントをde novoに(罹患者で起こった新生変異として)持っており、家族歴がある場合には、罹患した両親の1人から受け継いでいた。
疾患名
1970年代前半に最初にこの疾患が報告されてから、SHORT症候群と思われる疾患がさまざまな異なる名称で報告されてきた。
- 低出生体重のRieger症候群
- Rieger奇形、低身長、インスリン不足型糖尿病を伴う常染色体性部分型脂肪萎縮症*
- 痩せ型、小顔面骨の無虹彩症*
* 2番目と3番目の病名で報告された患者は、後にPIK3R1の病的バリアントを持ち、SHORT症候群であることが示された。
頻度
SHORT症候群は非常に稀な疾患である。文献で報告されているのは50例に満たない。
人種による頻度の違いは知られていない。
遺伝学的関連(アレル)のある疾患
生殖細胞系列にPIK3R1遺伝子の病原性バリアントを持つ2人の患者で、異なる表現形が報告されている。
- 1人はPIK3R1遺伝子のエクソン6に早期ストップコドンをホモ接合性に有していた。このためp85αアイソフォームが欠損し(ほかのアイソフォーム[p55α、p50α]は作られる)、無ガンマグロブリン血症を呈した[Conley et al 2012]。「分子遺伝学」の項参照。
- もう1人はPIK3R1タンパク質のN末端側SH2ドメインのアミノ酸置換を持ち、黒色表皮腫と重度のインスリン抵抗性を呈した [Baynes et al 2000]。
SHORT症候群の症状のない人に発生する単発の腫瘍が、PIK3R1遺伝子の体細胞バリアントを有する可能性がある。詳細は「分子遺伝学 がんと良性腫瘍」の項を参照。
鑑別診断
子宮内胎児発育遅延(IUGR)と出生後の成長障害がRussell-Silver症候群によくみられる特徴である。Russell-Silver症候群の患者でも、三角形の顔など、顔つきの特徴が共通してみられることがある。Russell-Silver症候群は、遺伝学的には不均質な疾患で、父親由来の染色体11p15にあるインプリンティング・センター1(IC1)の低メチル化によるものが35-50%、7番染色体の母親由来の片親性ダイソミー(UPD)が10%の患者でみられる。Russell-Silver症候群と臨床診断されても分子遺伝学的異常が検出されない患者がSHORT症候群と診断されることもあるので、このような患者では顔の表現形を注意深く検討することが望まれる。低身長は、Russell-Silver症候群の方がSHORT症候群よりも顕著である。前眼房奇形や脂肪萎縮症などSHORT症候群でみられるその他の特徴は、Russell-Silver症候群では一般的ではない。
Alagille症候群は、肝臓、心臓、眼、顔、骨格といった多臓器が関連する複雑な疾患である。遺伝形式は常染色体優性である。JAG1またはNOTCH2遺伝子の病的バリアントをヘテロ接合性に持つこと、またはコピー数変異が原因で起こる。Alagille症候群とSHORT症候群の眼と歯の所見は同様の場合があるが、Alagille症候群の特徴である肝臓疾患は、SHORT症候群では報告されていない。
前眼房奇形
非症候性の前眼房奇形は、PITX2(OMIM 601542)とFOXC1(OMIM 601090)を含むいくつかの遺伝子と関連している。これら2つの遺伝子バリアントによる非症候性の前眼房奇形は、常染色体優性形式で遺伝する。
PITX2(4q25)とBMP4(14q22)を含む領域のコピー数バリアント(染色体マイクロアレイ検査でしばしば検出される)は、SHORT症候群とは臨床的に異なる症候性の前眼房奇形を起こすことがある [Karadeniz et al 2004, Lines et al 2004, Reis et al 2011]。
Berardinelli-Seip先天性脂肪萎縮症は、出生時か出生後早期に診断されることが多く、重度かつ全身性の脂肪萎縮、肝腫大、末端肥大症様顔貌を特徴とする。脂肪萎縮症の程度、それに関連する顔の特徴が、SHORT症候群とは異なっている。SHORT症候群と同様に、インスリン抵抗性と糖尿病が、思春期後半から成人早期に起こってくる。Berardinelli-Seip先天性脂肪萎縮症は、常染色体劣性遺伝形式の疾患で、AGPATまたはBSCL2遺伝子どちらかの、両アレルの変異が原因である。
Hutchinson-Gilford早老症候群
一般的にこの症候群は、LMNA遺伝子のde novoの病的バリアントをヘテロ接合性に持つことで起こる。臨床症状は小児期にあらわれ、疾患の進行とともに老化が進むという特徴は、SHORT症候群とは性質の異なるものである。これに対してSHORT症候群の患者では、一般に、形質的特徴(脂肪萎縮症と未発達な鼻翼など)が出生時に明らかである。SHORT症候群の顔の特徴は、Hutchinson-Gilford早老症のほかJohansson-Blizzard症候群(UBR1遺伝子の両アレルの病原性バリアントによる)やHallerman-Streiff(遺伝的原因はわかっていない)と間違えられることがある。
臨床的マネジメント
初回診断後の評価
SHORT症候群と診断された患者の疾患の程度とニーズを明らかにするために、初回診断時に行っていなければ、以下の評価を行うことが勧められる。
SHORT症候群と診断された直後の患者(年齢は問わない)に対して
- 眼の発生障害や緑内障の管理経験を有する眼科医による前眼房と眼圧の診察
- 聴力評価
- 臨床遺伝専門医の診察
SHORT症候群と診断された乳児に対して
- 身長と体重の測定
- 発達遅滞がある場合、言語発達または運動発達の評価
小児期後半および成人でSHORT症候群と診断された患者に対して
- 歯科検診
- インスリン抵抗性/糖尿病の評価
症状の治療
Axenfeld-Rieger奇形または関連する前眼房奇形
眼の発生障害や緑内障の管理経験を有する眼科医によって、眼圧を低下させ、安定させることで視力を温存するための治療を行う。
歯科的所見
歯科治療を行う。歯冠や歯科補綴物を使用することもある。
インスリン抵抗性/糖尿病
耐糖能障害と糖尿病に対し、食事療法、生活スタイル改善、経口薬、インスリンを使った標準治療を糖尿病専門医の管理下で行うことが推奨される。
感音性難聴
「難聴・遺伝性難聴概説」を参照されたい。
サーベイランス
以下のサーベイランスが適切である。
- 身長、体重、BMIを含む成長の定期的なモニタリング
- 前眼房奇形のある患者では、眼の発生障害や緑内障の経験を有する眼科医による継続的な眼科検診
- インスリン抵抗性と糖尿病のスクリーニングを小児期中期に開始
- 小児期には定期的な聴力評価
回避すべき薬剤/環境
成長ホルモンを使用した患者ではインスリン抵抗性のリスクが増加するため、SHORT症候群では成長ホルモンは禁忌と考えられる[Thauvin-Robinet et al 2013]。
血縁者のリスクの評価
遺伝カウンセリングの目的については、「遺伝カウンセリング」の項で、リスクのある血縁者の検査に関連した問題を参照されたい。
妊娠管理
SHORT症候群の女性の妊娠・出産例がある。糖尿病がある場合は、適切な管理を行う。
SHORT症候群によってIUGRが起こるリスクが50%あることを踏まえて、胎児の成長をモニターすべきである。
研究中の治療
ClinicalTrials.govでは、幅広い疾患や状況の臨床研究の情報が得られる。
注:SHORT症候群の臨床試験は登録されていないかも知れない。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
SHORT症候群は常染色体優性遺伝である。
患者家族のリスク
発端者の両親
- SHORT症候群と診断された患者の一部は罹患した両親を持つ。
- 発端者で発見された病的バリアントが両親どちらかの白血球由来DNAで検出されない場合は、両親が性腺モザイクである場合と、発端者で起こった新生突然変異の2つの可能性がある。性腺モザイクが確定した例の報告はないが、可能性としては残る。特記事項として、Gorlin et al [1975]が1975年に報告したSHORT症候群の同胞発症例は、性腺モザイクが原因であると思われる。
- 新生突然変異が原因の患者の割合は知られていない。これまでの報告例では、8/12が新生突然変異とされるが、ほかの報告にあるSHORT症候群の孤発例の両親では、発端者の病原性バリアントが新生突然変異かどうかの評価が十分に行われていないため、少なく見積もられている可能性がある。
- 両親の評価により、表現形が軽症のために今まで診断されなかった人が見つかることがある。従って、適切な評価を行うまで、完全に家族歴がないとは確定できない。これまで、全ての罹患者の状態は、医師の診察(低身長、脂肪萎縮症、特徴的な顔立ちの確認が基本)により正しく確認されている。 発端者の同胞
- これまで検査を受けた罹患男性の母親すべてが保因者であるという観察から,疾患原因CAGリピートアレルが伝わるリスクは各妊娠で50%である.
- 疾患原因CAGリピートアレルを受け継いだ男性の同胞は発症する:アレルを受け継いだ女性の同胞は保因者となるが通常は発症しない.
発端者の同胞
発端者の同胞のリスクは、発端者の両親の遺伝学的状況によって異なる。
- 発端者の両親のどちらかが罹患している場合、同胞のリスクは50%である
- 両親が臨床的に罹患していない時には、発端者の同胞のリスクは低いと思われる。両親が罹患していない場合の同胞の再発率は、明らかではない。
- 両親が臨床的に罹患していない発端者の同胞は、両親での浸透率が低い可能性が理論上あるため、SHORT症候群のリスクは一般より高い。
- 発端者が持つ病原性バリアントが両親の白血球由来DNAで検出できない場合、同胞のリスクは低いが、性腺モザイクの可能性があるため、一般よりは高い。
発端者の子
SHORT症候群の患者の子供には、50%の確率で病原性バリアントが受け継がれる。
その他の血縁者
その他の血縁者のリスクは、発端者の両親の状況による。両親のどちらかが罹患していれば、その血縁者にはリスクがある。
遺伝カウンセリングに関連した問題
新生変異の病原性バリアントと思われる家族で考慮すべきこと
常染色体優性遺伝疾患の発端者の両親が病的バリアントを持たない、あるいは疾患の臨床的証拠を有さない場合、発端者の変異は新生変異である可能性が高い。しかし、生殖補助医療により父親または母親との血縁関係がない場合や、明らかにされていない養子縁組などの非医学的な説明の可能性も考慮される。
家族計画
- 遺伝学的リスクの算定および出生前診断の可能性についての話し合いは、妊娠前が最適である。
- 罹患している若年成人やリスクのある若年成人に対して、遺伝カウンセリング(子の潜在的リスクや生殖手段についての話し合いを含む)を提供することが適切である。
DNAバンキング
DNAバンキングは、将来使用する可能性に備えてDNA(一般に白血球細胞から抽出したもの)を保存しておくことである。検査方法、および我々の遺伝子、アレルバリアント、疾患に関する理解は将来進歩する可能性があるため、罹患者のDNAバンキングについて考慮すべきである。
出生前診断
家系内でPIK3R1遺伝子の病原性バリアントが同定されている場合、この疾患/遺伝子の検査が可能な、または依頼に応じられる臨床検査機関によって、リスクの高い妊娠に対する出生前診断が可能である。
SHORT症候群のように、知的障害がなく、治療可能な疾患に対する出生前診断の希望は一般的ではない。出生前診断を行うかどうかについて、特に早期診断のためでなく妊娠中断を目的に考慮される場合には、医療専門家の間でも家族内でも、考え方に違いがある可能性がある。多くの医療機関では、出生前診断は両親の選択に沿って実施を検討するが、このような問題について話し合うことが適切である。
着床前診断(PGD)は、家系内でPIK3R1遺伝子の病的バリアントが同定されていれば、選択肢の一つとなり得る。
分子遺伝学
分子遺伝学およびOMIMの表の情報は、GeneReviewsの他の部分の記載内容と異なる可能性がある。表には、より最近の情報が含まれていることがある。
表A
SHORT症候群:遺伝子およびデータベース
| 遺伝子 | 染色体座位 | タンパク質 | 遺伝子特異的データベース | HGMD |
|---|---|---|---|---|
| PIK3R1 | 5q13?.1 | Phosphatidylinositol 3-kinase regulatory subunit alpha | PIK3R1base: Database for pathogenic mutations in the p85-alpha SH2 domain p85αSH2ドメインの病原性バリアントデータベース |
PIK3R1 |
データは以下の標準的参照資料をもとに作成した。遺伝子はHGNC、染色体座位、座位の名称、遺伝子変異に密接に関連した領域、相補群はOMIM、タンパク質はUniProtを参照した。遺伝子特異的データベースとHGMDの説明は、こちらを参照されたい。
表B
SHORT症候群のOMIM 登録番号(内容はOMIMを参照されたい)
| 171833 | PHOSPHATIDYLINOSITOL 3-KINASE, REGULATORY SUBUNIT 1; PIK3R1 |
| 269880 | SHORT SYNDROME |
遺伝子構造
PIK3R1遺伝子の最も長い転写産物(NM_181523.2)は、16のエクソンを持ち、ゲノム長は86kb以上である。加えて、選択的スプライシングによる3つの転写バリアントが知られている(NM_181524.1, NM_181504.3, NM_001242466.1)。NM_181523.2は724個のアミノ酸からなるタンパク質(p85αと呼ばれる)をコードする。転写バリアントとアイソフォームの詳細については、表Aの「遺伝子」のリンクを参照。
病原性アレルバリアント
これまでに知られている病原性バリアントには、微小欠失、ミスセンス変異、ナンセンス変異がある。これらの大部分は、PI3Kの活性調節部位である3'側src相同配列ドメイン2(SH2)にみられる。
SHORT症候群の16家系中10家系では、頻度の高い病原性バリアントc.1945C>Tが保有されている(表1参照)
c.1615_1617delATTとc.1465G>Aの2つの病原性バリアントは、p110触媒サブユニットの活性調節部位で起こったものである[Thauvin-Robinet et al 2013]。
PIK3R1のその他のバリアントは、SHORT症候群を起こさず、違った表現形に関連する(「遺伝学的に関連がある疾患」の項参照)。
表2
PIK3R1遺伝子の病原性アレルバリアント
| DNAの塩基変化 | タンパク質のアミノ酸変化 | 参照配列 |
|---|---|---|
| c.1945C>T 1 | p.Arg649Trp | NM_181523.2 NP_852664.1 |
| c.1906_1907delAA | p.Asn636ProfsTer17 | |
| c.1971T>G | p.Tyr657Ter | |
| c.1615_1617delATT | p.Ile539del | |
| c.1465G>A | p.Glu489Lys | |
| c.1943dupT | p.Arg649ProfsTer5 | |
| c.1892G>A | p.Arg631Gln |
バリアントの分類に関する注記:表中のバリアントはそれぞれの著者が報告したものである。GeneReviewsのスタッフは、バリアントの分類を独立した立場で検証していない。
命名法に関する注記:GeneReviewは、HGVS(www.hgvs.org)の標準的な命名法に従っている。命名法の説明は、「Quick Reference」を参照されたい。
- 頻度の高い変異;表1参照。
正常な遺伝子産物
この遺伝子は、PI3K(フォスファチジルイノシトール3リン酸化酵素)の調節サブユニットαをコードし、触媒サブユニットであるp110を安定化させる。調節サブユニットp85αにリン酸化されたチロシン残基が結合すると、p85α/p110αヘテロマーの構造に変化が起こってp110の阻害が外れる。
PIK3R1の正常遺伝子産物には85、55、50キロダルトンの3つのアイソフォームがある。主要なアイソフォーム85αは、ほかのアイソフォームがそれぞれ骨格と肝臓で優勢であるのに対し、広範に発現している。85αアイソフォームは、4つの機能ドメイン(SH3ドメインとRho-GAPドメインが各1、SH2ドメインが2)を持つ。p85α/p110αヘテロマーPI3Kは、フォスファチジルイノシトール(3,4)2リン酸をフォスファチジルイノシトール(3,4,5)3リン酸(PIP3)に変える。PIP3はAKTを誘導し、下流で起こる細胞成長に関わるイベントを開始させる。転写バリアントの詳細は、表A「遺伝子」を参照。
変異遺伝子産物
変異遺伝子産物は、ハプロ不全のメカニズムにより、SHORT症候群を起こすと思われるが、メカニズムに関する研究は少ない。
- p.Asn636ProfsTer17短縮型バリアントを持つリンパ球細胞株では、AKT-mTOR経路の下流でリン酸化の減少がみられる[Dyment et al 2013]。
- PIK3R1の病原性バリアントを有する患者の線維芽細胞では、AKT経路活性化能とグルコース取り込み能の減少がみられ、表現形として重度のインスリン抵抗性が予想された [Thauvin-Robinet et al 2013]。
- これらの線維芽細胞では、インスリン受容体基質(IRS1)との相互作用が顕著に低下していた。インスリン刺激によるAKTの活性化は、脂肪細胞でも減少していた [Chudasama et al 2013]。
がんおよび良性腫瘍
単独の散発性腫瘍(神経膠芽腫、乳がん、子宮内膜がん、膀胱がん、尿路上皮がん、卵巣がん、大腸がん、胃がんなど)が体細胞性のPIK3R1バリアントを有する可能性がある。SHORT症候群の症状がない場合には生殖細胞系列のバリアントではないため、腫瘍の起こりやすい体質が遺伝性することはない。
更新履歴
- Gene Reviews著者: A Micheil Innes, MD, FRCPC, FCCMG and David A Dyment, DPhil, MD, FRCPC, FCCMG.
日本語訳者: 菅原宏美 田村和朗(近畿大学大学院総合理工学研究科 理学専攻 遺伝カウンセラー養成課程)
Gene Reviews 最終更新日: 2014.5.15.日本語訳最終更新日: 2017.8.10.[in present]
![]()