APC関連ポリポーシス
(APC-Associated Polyposis Conditions)
[Synonyms: SCA2]
Gene Reviews著者: Timothy Yen, MD, Peter P Stanich, MD, Lisen Axell, MS, CGC, and Swati G Patel, MD, MS.
日本語訳者: 箕浦祐子(札幌医科大学大学院医学研究科遺伝医学),櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.5.12. 日本語訳最終更新日: 2022.6.8
原文 APC-Associated Polyposis Conditions
要約
疾患の特徴
APC関連ポリポーシスには,(古典型または軽症型)家族性大腸ポリポーシス(FAP),胃腺がんおよび胃近位ポリポーシス(GAPPS) が含まれる.
- FAPは大腸がん易罹患性症候群であり,古典型と軽症型がある.古典型FAPは数百~数千個の大腸腺腫性ポリープを特徴とし,発症平均年齢は16歳(7-36歳)である.
- 古典型FAPでは,35歳までに95%の患者にポリープが発生する;大腸切除術を施行しない限り大腸がんは必発である.未治療患者における大腸がん診断の平均年齢は39歳(34-43歳)である.軽症型の特徴は,複数の大腸ポリープ(平均30個)が生じ,より近位で発生すること,古典型FAPよりも高齢になってから大腸がんと診断される点である.
- 軽症型FAPでは,大腸がんの生涯リスクは70%で,診断年齢中央値は50~55歳である.大腸外病変は,胃・十二指腸ポリープ,骨腫,歯牙異常,先天性網膜色素上皮肥大(congenital hypertrophy of the retinal pigment epithelium: CHRPE),良性の皮膚病変,デスモイド腫瘍,副腎腫瘍やその他関連がんなど,多岐にわたる.
- GAPPSは, 胃近位ポリポーシスを特徴とし,胃がんのリスクが高まる.十二指腸や大腸との関連はほとんどの患者で報告されていない.
診断・検査
APC関連ポリポーシスは,APC遺伝子にヘテロ接合型生殖細胞系列病的バリアントが同定されることによって確定する.
臨床的マネジメント
症状の治療:
大腸サーベイランスで見つかった5mmよりも大きいポリープは全て摘除する.大腸がんの診断または疑いがある,あるいは重大な症状(出血/閉塞)がある場合は,大腸切除術の絶対的適応となる.内視鏡的切除の適応とならない10mmよりも大きな複数の腺腫がある場合や,サーベイランス期間中に顕著な腺腫数の増加が認められた場合,高悪性度異形成を伴う腺腫がある場合,大腸を正確に調べられない状況(例えば,無数の小型腺腫の存在,あるいは大腸内視鏡のアクセスや適応が限られている場合)などは,大腸切除術の相対的適応となる.十二指腸腺腫は,絨毛性変化や高度異形成を生じた場合,直径が1cmを超えた場合,あるいはSpigelman分類のstageが進行した場合には,内視鏡あるいは外科的切除が考慮される.上部消化管内視鏡検査で進行した胃の病変が見つかった場合は,胃切除術を考慮する.骨腫は整容的な理由で切除することがある.デスモイド腫瘍は外科的に切除されるか,進行している場合は非ステロイド消炎鎮痛薬(NSAIDs),抗エストロゲン薬,細胞障害性化学療法,および/または放射線照射によって治療する.副腎腫瘍と甲状腺がんについては,必要に応じて標準治療を行う.数々の研究で,NSAIDsおよびエルロチニブがFAP患者における腺腫を退縮させ,ポリープによる症状を減らすということが示されたが,その後のがんリスクへの影響が不明であることから、現在FDAが承認しているFAPのための化学予防薬は存在しない.
一次症状の予防:
古典型FAP患者では,大腸全摘術は大腸がんのリスクを減少させる.軽症型FAP患者では,大腸全摘術の適応とはなるが,約1/3の患者では,定期的に大腸ポリープ切除術を伴うサーベイランスを行うことで,大腸がんを十分に予防できる程度の数の大腸ポリープしか発生しない.GAPPSの患者に対して予防的胃切除術を考慮するべきかどうかは,現段階では不明である.
サーベイランス:
大腸内視鏡によるスクリーニングについては,古典型FAPについては10-15歳から,軽症型FAPについては青年期後半から開始する.20-25歳あるいは大腸手術前には,Vater膨大部の可視化を伴う食道胃十二指腸鏡検査,Spigelman分類のstageが進行している場合は,全小腸の可視化も考慮する.1年ごとに甲状腺の触診,甲状腺超音波検査,神経学的検査,および(デスモイドに対する)腹部検査を実施する.肝芽腫のスクリーニングには,3~6ヶ月ごとに,肝臓の触診,肝超音波検査と血清αフェトプロテイン濃度の測定を,5歳まで行う.GAPPS患者に対する胃がんスクリーニングの効果については,現段階では不明である.
避けるべき薬剤や環境:
デスモイドのリスクの高い人に対する複数回の手術;出産前の女性に対する大腸全摘・回腸嚢肛門吻合術(IPAA).
リスクのある親族の検査:
分子遺伝学的検査によって早期にリスクのある血縁者を診断することは,診断の確実性を上げ,病的バリアントを受け継いでいない血縁者に対して経済的負担のかかるスクリーニングを回避することができる.
遺伝カウンセリング
APC関連ポリポーシスは常染色体顕性遺伝(優性遺伝)の形式をとる.約75-80%の患者には罹患した親がいる.患者の子は50%の確率でAPC遺伝子の病的バリアントを受け継ぐ.家系内の発端者において病的バリアントが同定されている場合には,出生前診断や着床前診断が可能である.
訳注:日本では,本症に対する出生前診断や着床前診断は行われない.いずれにしても次世代への遺伝に関しては細心の遺伝カウンセリングが必要である.本稿の記載範囲
APC関連ポリポーシス には以下の病態が含まれる.1
- 家族性大腸ポリポーシス(FAP)
- 軽症型FAP
- 胃腺がんおよび胃近位ポリポーシス(GAPPS)
同義語と旧名については,病名を参照すること.
- これらの表現型のその他の遺伝的要因については鑑別診断の項を参照.
診断
APC関連ポリポーシスが疑われる所見
米国総合癌ネットワーク(NCCN)は家族性大腸ポリポーシス(FAP)および軽症型FAPの診断を考慮する際のアルゴリズムを発表している[Weiss et al 2021] (全文はこちら).これらのガイドラインでは,APC遺伝子の遺伝学的検査が推奨されている.GAPPSの診断ための合意の得られたガイドラインはまだ存在しない.
NCCNガイドラインでは,以下の臨床的所見が見られる場合,APC関連ポリポーシスを疑うべきであるとされている.
- 複数の大腸腺腫性ポリープ(少なくとも累計10-20個)
- 大腸腺腫性ポリープの家族歴(10個を超えるポリープがある人が1人,または複数のポリープがある近親者が2人以上いればそれ以下の個数でも考慮する.特に若年発症の場合),既知のAPC病的バリアント,および/または大腸以外のAPC関連ポリポーシスの所見が見られる場合.
- 肝芽腫
- 多発/両側性の先天性網膜色素上皮肥大(CHRPE)
- デスモイド腫瘍
- 篩型甲状腺乳頭がん
上記に加え,APC関連ポリポーシスが疑われる特徴.腺腫性ポリープが少数~ない状態での大腸がんの若年発症,歯牙異常(過剰歯など),骨腫,歯牙腫,類表皮嚢胞,十二指腸腺腫およびがん,胃底腺ポリポーシス,胃がん,膵がん,小腸がんおよび/または髄芽腫.
確定診断
APC関連ポリポーシスの診断は,APC遺伝子にヘテロ接合型生殖細胞系列病的バリアント(pathogenic variant)(またはおそらく病的バリアント(likely pathogenic variant))が同定されることによって確定する(Table 1参照)
注:ACMGのバリアント解釈のためのガイドラインでは,“病的バリアント”および“おそらく病的バリアント”は臨床上は同義として扱っており,どちらも診断に役立つと考えられ,臨床的な意思決定に使用することが可能である.この項で“病的バリアント”と表記している場合は,“おそらく病的バリアント”も含むと理解されたい.
古典型FAPの診断は,分子遺伝学検査によって APC遺伝子にヘテロ接合型生殖細胞系列病的バリアントを認め,以下の条件のいずれかを満たす場合になされる.
- 100個以上の大腸腺腫性ポリープ (若年発症者や大腸切除術を実施した人については,腺腫性ポリープが100個未満の場合も).
- 腺腫性ポリープは100個未満であるが,血縁者が古典型FAPと確定診断されている.
軽症型FAP(attenuated FAP)の診断は,分子遺伝学的検査でAPC遺伝子にヘテロ接合型生殖細胞系列病的バリアントがあり,以下の条件に合致する場合に考慮される.
- 血縁者に軽症型FAPと確定診断された患者がいる;そして/または
- 大腸の腺腫性ポリープが100個未満;あるいは
- 40歳を超えた時点で,大腸の腺腫性ポリープが100個を超えた.
GAPPSの診断は,分子遺伝学的検査でAPC遺伝子のプロモーター1B領域にヘテロ接合型生殖細胞系列病的バリアントがあり,以下の条件を満たす場合になされる.
- 胃体部および胃底腺の限局性ポリープ
- 胃の近位に100個より多いポリープがあるか,あるいは第1度近親者にGAPPSの患者がおり,30個より多いポリープがある.
- 主に胃底腺ポリープ(FGPs)といくつかの胃腺腫;異形成の領域を有するもの(または異形成性FGPまたは胃腺がんの家族歴がある).
- 大腸あるいは十二指腸に多発ポリープの形跡がない.
分子遺伝学的検査
- 分子遺伝学的検査には,単一遺伝子検査およびマルチジーンパネルを用いる方法がある.
- 単一遺伝子検査.APC遺伝子の配列解析および欠失/重複解析の両方を実施するべきである.最初の検査でAPC遺伝子に病的バリアントが見つからない場合は,欠失/重複解析で制御領域(特にプロモーター1B)の解析も実施する.
注:APC関連ポリポーシスが疑われ,生殖細胞系列の検査が陰性だった場合,腺腫組織のAPC遺伝子配列解析および欠失/重複解析も考慮されうる.生殖細胞系列の検査が陰性で複数の腺腫に同じAPC遺伝子病的バリアントが同定されれば,体細胞モザイクとみなされる [Aretz et al 2007].孤発例の約20%がAPC遺伝子病的バリアントの体細胞モザイクである.
- APC遺伝子や他の関連遺伝子(鑑別診断の項を参照)を含むマルチジーンパネルも考慮する.注:(1)パネルに含まれる遺伝子や検査の精度は,検査機関によって異なっているだけでなく,時代とともに変化する.(2)マルチジーンパネルには本稿で扱っている病態に関連のない遺伝子が含まれている場合もあるため,臨床医はどのマルチジーンパネルが症状の遺伝的要因を特定する可能性が最も高いかを決定する必要があり,同時に,意義不明なバリアント(VUS)や,根本的な表現型を説明しえない遺伝子における病的バリアントの同定を避ける必要がある.(3)検査機関によっては,臨床医が指定した遺伝子を含むカスタム設計された検査機関独自のパネルや,表現型に焦点を当てたエクソーム解析パネルといった選択肢が存在することもある.(4)こうした検査で用いられる手法には,配列解析,欠失/重複解析,配列解析以外の検査がある.
マルチジーンパネルの概要についてはこちらをクリック.臨床医のための,遺伝学的検査依頼に関するより詳細な情報は こちら を参照.
表 1. APC関連ポリポーシスで使用される分子遺伝学的検査
| 遺伝子¹ | 検査方法 | この方法により検出可能な病的バリアント2を有する発端者の割合 |
|---|---|---|
| APC | 配列解析3 | £90%4,5 |
| 欠失/重複解析6 | 8-12%4 |
- 染色体座位およびタンパク名はTable A.遺伝子とデータベースの項を参照のこと.
- この遺伝子で検出されるバリアントの情報は分子遺伝学の項を参照のこと.
- 配列解析で検出される変異は非病的バリアント,おそらく非病的バリアント,意義不明なバリアント,おそらく病的バリアント,病的バリアントがある.バリアントには小規模な遺伝子内欠失/挿入やミスセンス,ナンセンス,スプライス部位バリアントが含まれる.通常,エクソンや全遺伝子の欠失/重複は検出されない.配列解析結果に対する解釈はこちらを参照のこと.
- ヒト遺伝子変異データベース(HGMD) Professional(定額制サイト)からのデータ [Stenson et al 2020]
- 孤発例の約20%が体細胞モザイクであり[Hes et al 2008],リンパ球から抽出したDNAを用いた分子遺伝学的検査では,APC病的バリアントを検出できない可能性がある [Aretz et al 2007, Hes et al 2008].
- 標的遺伝子の欠失・重複解析では,遺伝子内の欠失や重複が検出できる.検査方法は,定量的PCR,ロングレンジPCR,MLPA(multiplex ligation-dependent probe amplification)法,単一エクソンの欠失や重複の検出を目的とする標的遺伝子マイクロアレイなどである.大規模な欠失/重複の検査には,APC遺伝子の制御領域(特にプロモーター1B)が含まれるべきである[Rohlin et al 2011].
疾患の特徴
臨床症状
APC関連ポリポーシスには古典型家族性大腸ポリポーシス (FAP),軽症型FAP,および胃腺がんおよび胃近位ポリポーシス (GAPPS)が含まれる.
FAP
古典型FAPの患者は大腸腺腫性ポリープが10~20歳代に出現しはじめる;ポリープが診断される平均年齢は16歳(7-37歳)である [Petersen et al 1991].35歳までには95%のFAP患者でポリープが発生する.一度ポリープができるとその数は急速に増加し,病変が完全に進行すると典型例では数百から数千におよぶポリープが認められる.大腸切除術を行わなければ大腸がんは必発である.未治療患者が大腸がんと診断される平均年齢は39歳(34-43歳)である;未治療のFAP患者の大腸がんを発症するリスクは21歳までで7%,45歳までで87%,50歳までで93%である.稀ではあるが,50歳台で無症状の患者も報告されている.家系間,家系内で表現型が様々に異なる [Half et al 2009].
軽症型FAPは,古典型FAPより少ない大腸ポリープ(平均30個)を特徴とするが,大腸がんのリスクは高い. Knudsen et al [2010]による国際共同研究では,軽症型FAPは25歳以上で100個以下の大腸腺腫性ポリープを有するものと臨床的に定義された.古典型FAPよりも大腸の近位でポリープが発生する傾向がある.軽症型FAP患者が大腸がんと診断される平均年齢は50-55歳であり,古典型FAPよりも15年ほど遅いが,散発性の大腸がんよりは若い [Spirio et al 1993, Giardiello et al 1997].軽症型FAPの80歳までの大腸がん累積リスクは70%と推定される [Neklason et al 2008].
FAPに認められるその他の特徴
表2. 家族性大腸ポリポーシスにおける大腸以外のがんの生涯発症リスク
| 部位 | がんのタイプ | がんの生涯リスク |
|---|---|---|
| 小腸:十二指腸(多くは乳頭部周囲) | 腺がん | 4%-12% |
| 小腸:十二指腸より遠位部 | がん | 稀 |
| 膵臓 | 腺がん | ~1% |
| 甲状腺 | 甲状腺乳頭がん | 1-12% |
| 中枢神経 | 通常は髄芽腫 | <1% |
| 肝臓 | 肝芽腫 | 1.6% |
| 胆管 | 腺がん | 低いが,一般集団より高い |
| 胃 | 腺がん | 2016年では西洋文化圏において1.3% [Mankaney et al 2017] |
小腸ポリープおよび小腸がん.十二指腸の腺腫性ポリープはFAP患者の50-90%に認められ,通常十二指腸の第2部,第3部に発生する[Kadmon et al 2001].回腸や空腸での発生は多様である [Koornstra 2012].十二指腸ポリープはポリープの数と大きさ,組織像,異形成の程度による分類法が確立している [Spigelman et al 1989].大腸ポリープの数と上部消化管ポリープの数との明らかな相関は認められない[Kadmon et al 2001].
乳頭部周辺(十二指腸乳頭部とVarter膨大部を含む)の腺腫性ポリープは,内視鏡的に見えづらいあるいは見えないものもあるが,少なくとも50%のFAP患者に認められる [Mehta et al 2021].この領域のポリープは膵管の閉塞をきたす結果,膵炎または胆道閉塞を起こしうるため,FAPではこれらの合併症の頻度が高い.こうしたポリープは小さいことが多く,可視化するために先端キャップのついた胃内視鏡や側視鏡による検査が必要となる [Yang et al 2020].膵胆管系の分泌物(胆汁など)がこの部位の腺腫の発生に影響するという説があり [Wallace & Phillips 1998] ,膨大部周辺のポリープの悪性化リスクの上昇の理由である可能性がある [Kadmon et al 2001].
小腸がんの生涯発症リスクは4-12%で,大多数は十二指腸に発生する.十二指腸における腺がんは膨大部に最も多く見られる.17~81歳の間に発症し,診断時年齢中央値は45~52歳と報告されている [Wallace & Phillips 1998, Kadmon et al 2001]. 小腸がんは十二指腸遠位部にも発生するが,稀である. Ruys et al [2010] は,FAP患者において,空腸がん17例,回腸がん3例の報告があったとしている.
膵がん.限定的なデータしかないが,197のFAP家系に関する研究では,FAP患者およびリスクのある血縁者において,膵がんの相対的リスクは一般集団より4.5倍高いことが示されている.Giardiello et al [1993b] は,FAP患者における80歳までの膵がんの生涯発症リスクは1%と推定している.この報告以降のFAP患者における膵臓癌の症例報告はわずかである[Moussata et al 2015].
甲状腺がんおよび良性甲状腺疾患.FAP患者における甲状腺がんの頻度は報告によって非常にばらつきがある.様々な後ろ向き調査を取りまとめた総説では0.4-2.6%である一方で,前向き研究では有病率がより高く,2.6%-11.8%と報告されている [Cetta 2015, Chenbhanich et al 2019].FAP患者における甲状腺がんの頻度は,女性:男性比が80:1と著しい性差があり,80%以上の患者が18~35歳の間に診断される [Cetta 2015].甲状腺乳頭がんの稀な亜型である,篩型バリアントはFAPと関連するが [Pradhan et al 2015],散発性のがんでも起こりうる.
FAP患者の良性甲状腺疾患の頻度に関するデータは限定的である.システマティックレビューおよびメタ解析では,FAP患者の6.9%が甲状腺の内分泌疾患(甲状腺機能低下症,甲状腺腫および/または甲状腺炎)を有しており,48.8%に良性の甲状腺結節があった [Chenbhanich et al 2019].研究間で結果が著しく異なるため,甲状腺疾患の割合に矛盾がみられるものと思われる.家族集積性と女性優位性が見られる.
中枢神経系(CNS).FAP患者にみられる最も一般的なCNS腫瘍は髄芽腫である.CNS腫瘍のリスクがFAP患者において増加するのは事実であるが,その絶対的リスクは約1%にすぎない [Attard et al 2007].
肝芽腫.FAP患者における肝芽腫のリスクは一般集団より750~7,500倍も高いが,その絶対的リスクは2%未満である.肝芽腫の大多数は3歳までに発生する [Aretz et al 2007].
胃ポリープおよび胃がん.FAPにおいては,胃底腺および腺腫性ポリープのリスクが高まる [Bülow et al 1995].胃底腺ポリープ(FGPs)は胃底部あるいは胃体部に発生する良性腫瘍である;これらを過誤腫様腫瘍と分類する人もいるが,この分類については未だ議論の下にある.FGPsはFAP患者の約半数に認められ,散発性のFGPsよりも異形成変化を生じることが多い[Bianchi et al 2008].FAPでは,腺腫性ポリープ(幽門腺腺腫,管状腺腫など) や過形成ポリープも認められる.
西洋文化圏に生活するFAP患者の胃がんのリスクは低いが,近年上昇傾向にある[Mankaney et al 2017].日本や韓国のFAP患者においては一般集団より10倍高い[Garrean et al 2008].胃腺がんはほとんどの場合,腺腫から発生すると考えられるが,FGPsから発生することもある[Attard et al 2001].内視鏡による高リスクポリープの同定を支援するための分類システムが提案されている [Mankaney et al 2020].
FAPにおける消化管以外の非悪性病変
骨腫はFAP患者の60-80%に発生する.骨の増殖が頭蓋や下顎骨に最も高頻度に見られるが,全身のどの骨にも生じうる.骨腫は通常臨床的に問題はなく,悪性化もしない;小児において大腸ポリープよりも先に現れることがある [Septer et al 2018].
歯牙異常.未萌出歯,1個以上の歯牙の先天的欠損,過剰歯,含歯性嚢胞(未萌出歯の歯冠に関連する歯原性嚢胞),歯牙腫などは,一般集団での発生頻度は1-2%のところ,FAP患者では約30-75%に出現すると報告されている. [Septer et al 2018].
先天性網膜色素上皮肥大 (CHRPE)は,網膜の不連続で平坦な色素性病変で,年齢とは関係なく生じ,臨床的に問題はない.CHRPE はFAP患者の80%に生じることが報告されている.CHRPEを可視化するには,散瞳させた上での間接検眼鏡による眼底検査を要する場合がある.リスクのある家系員で,多発性,または両側性のCHRPEが認められれば,FAPを受け継いでいる徴候である可能性があるが,単発性の病変は一般集団にも認められる [Rehan & Aye 2020].
良性の皮膚病変には類表皮嚢胞や線維腫が含まれ,顔面を含む全身に出現しうる.これらは悪性化する可能性はないので,主として整容上の問題となる.稀ではあるが,多発性毛母腫(良性の毛包腫瘍)も報告されている [Ciriacks et al 2020].
デスモイド腫瘍はFAP患者の10-30%に発症する [Nieuwenhuis et al 2011b, Sinha et al 2011].FAP患者でデスモイド腫瘍が発症するリスクは一般集団の800倍以上とされている.デスモイド型線維腫症患者の少なくとも7.5%はFAPである [Nieuwenhuis et al 2011a].よく分かっていないが,この良性の線維性腫瘍は筋線維芽細胞のクローン性増殖によるもので,局所的には浸潤性を有するが遠隔転移はしない.病理組織学的に異なる線維腫病変であるGardner関連線維腫は,デスモイド腫瘍の前駆病変であるとの仮説がある[Wehrli et al 2001].
デスモイド腫瘍の発生率は10~20歳代に最も高く,40歳までの発生が80%を占める[Sinha et al 2011].FAP患者のデスモイド腫瘍の約65%が腹部あるいは腹壁内に発生する[Sinha et al 2011]が,軸骨格や四肢にもよく生じる [Escobar et al 2012].デスモイド腫瘍が腹部臓器を圧迫したり,腹部手術を困難にすることもある.FAP患者の約5%がデスモイド腫瘍の発症および/または死亡を経験し,死亡率は腹腔内腫瘍で最も高いと報告されている[Sinha et al 2011].腹部のデスモイド腫瘍は自然に発生したり,手術後に発生したりする[Bertario et al 2001].妊娠によるデスモイド腫瘍の発生と進行への影響は不明である[Sinha et al 2011].デスモイド腫瘍発生の独立した予測因子としては:APC遺伝子コドン1399より3’側の病的バリアント,デスモイド腫瘍の家族歴,女性,腹部手術の既往があげられる[Sinha et al 2011].APC遺伝子5’側のコドン547-713領域の病的バリアントもデスモイド腫瘍と関連する[Slowik et al 2015].デスモイド腫瘍の家族歴は最もリスクへの影響が強く,第一度近親者にデスモイド腫瘍発症者がいれば,リスクは7倍となる[Sinha et al 2011].
MRIやCTスキャンがデスモイド腫瘍の評価に最も有用である[Escobar et al 2012].CTによるFAP患者のデスモイド腫瘍のスコアリングシステムが開発されている[Middleton et al 2003].
副腎腫瘍はFAP患者において,一般集団より2~4倍高いと報告されている [Rekik et al 2010].副腎腫瘍は一般集団の1-3%に認められるが,FAP患者では7.4-16%に認められた[Marchesa et al 1997, Shiroky et al 2018].107例のFAP患者による前向き研究では,13%に腹部CTで1cm以上の副腎腫瘍を認めた[Smith et al 2000b].ほとんどの副腎腫瘤は偶発的に見つかる非症候性の腺腫であるが,機能性病変や癌腫も生じうる[Marchesa et al 1997, Rekik et al 2010].
GAPPS
GAPPSは,胃近位FGPsと小腸型胃腺がんを特徴とし,通常明らかな十二指腸または大腸ポリープを認めない [Worthley et al 2012 ].GAPPS患者の胃がんの生涯リスクは13-25%である[Kim et al 2022].
遺伝型と表現型の相関
FAPの家系内・家系間の症状は一般的に多様であるが,遺伝型と表現型の相関に関する報告がいくつかある ( Table 3参照) .しかしながら,サーベイランスや予防的手術は罹患者の表現型に基づいて計画されるべきであり,遺伝型のみに基づくべきではない.同じバリアントを持つ患者の間でも,表現型が多様であるという研究報告も複数ある.
表現型の基準で軽症型FAPとされる患者の78%は,以下の3つの領域に病的バリアントを持つ:APC遺伝子5’側末端(コドン1-233),エクソン9の選択的スプライス領域(コドン311-412),3’側のコドン1595.しかし,遺伝型は必ずしも大腸の表現型を予測しえず;これら3領域のいずれかに病的バリアントを持つ患者では,一般的には軽症型FAPと関連しており,65%は100個以下の大腸腺腫を呈するが,35%は古典型FAPで大腸切除術を施行されている.大腸切除術の施行年齢中央値は18歳である[Anele et al 2022].
図1.
APC遺伝子の概略図
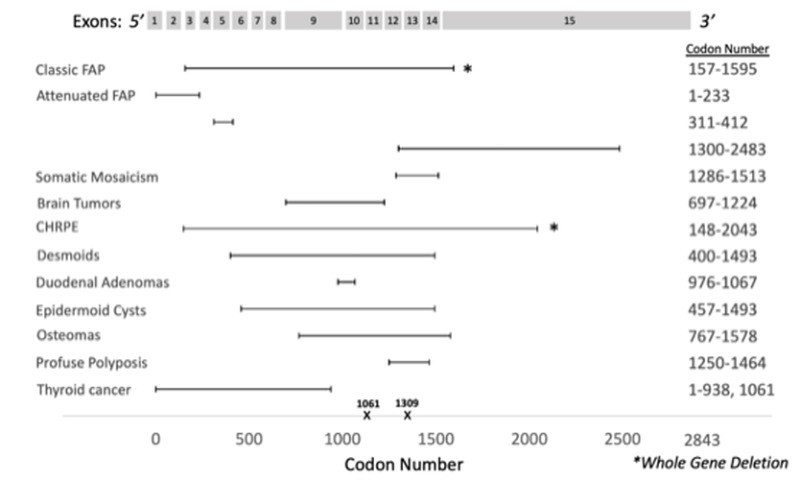
APC遺伝子は15のエクソンと2843のコドンで構成される.多くの生殖細胞系列バリアントは半分より5’側に位置し,特にコドン1061と1309に多い.遺伝子中央部分の生殖細胞系列バリアントは,古典型FAPとよく関連する.コドン1250~1464の間の生殖細胞系列バリアントは,密生型ポリポーシスと関連する一方,5’側または3’側末端は軽症型FAPと関連する.
図3.
APC関連疾患における遺伝型-表現型の相関
| 表現型 | 座位/APC 病的バリアントの種類 | 備考 | 参照 |
|---|---|---|---|
| FAP (古典型) | 全遺伝子欠失 | Quadri et al [2015] | |
| 軽症型FAP |
|
大腸外の病変 (CHRPEやデスモイド腫瘍など) は稀. | Sieber et al [2006], Anele et al [2022] |
| GAPPS | APC プロモーター1B | ||
| 体細胞モザイク | コドン1286-1513 (最も一般的) | 大腸の表現型:
|
Friedl & Aretz [2005], Hes et al [2008], Jansen & Goel [2020] |
| 脳腫瘍 | コドン697-1224 |
|
Attard et al [2007] |
| CHRPE | コドン148-2043 | Burger et al [2011] | |
| コドン311-1444 | 脚注2.参照 | ||
| 全遺伝子欠失 | Aretz et al [2005] | ||
| デスモイド | コドン1395-1493 | オッズ比: 4.37 | Sinha et al [2011] |
| 5'側のコドン400 | 発生率: 14.9% | Church et al [2015] | |
| コドン401-1400 | 発生率: 23.3% | ||
| 3' 側のコドン1400 | 発生率: 37.1% | ||
| 十二指腸腺腫 | コドン976-1067 | リスク4倍上昇 | Bertario et al [2003] |
| 類表皮嚢胞 | コドン457-1493 | Dinarvand et al [2019] | |
| 骨腫 | コドン767-1578 | ||
| 密生型ポリポーシス | コドン1250-1464 | ポリープの平均5000個 | D'Elia et al [2018] |
| 甲状腺がん | 5'側末端 (コドン1061またはコドン938より5’側) | Chenbhanich et al [2019] |
CHRPE=先天性網膜色素上皮肥大;FAP=家族性大腸ポリポーシス;GAPPS=胃腺がんおよび胃近位ポリポーシス
- 5'側のコドン233のバリアントは,軽症型FAPで最もよくみられるバリアントである [Knudsen et al 2003]
- ヒト遺伝子変異データベース(HGMD) Professional(定額制サイト)からのデータ [Stenson et al 2020]
浸透率
FAPの浸透率は100%である.
軽症型FAPでは,大腸ポリポーシスの浸透率はまだ完全に分かっていないが,80歳までの大腸がんの発生リスクはおよそ70%と推測されている[Neklason et al 2008].
GAPPSでは,FGPsおよび/またはintestinal-type(小腸型)胃腺がんの浸透率は不明である.
病名
FAPはしばしば100個以上の大腸ポリープがある古典的FAPを指す.古典的FAPとFAPは,同義として使われることもある.
Adenomatous polyposis coli(APC)は,歴史的にFAPを指す言葉として使われていたが,現在ではAPCは関連する遺伝子を指す.
APC関連ポリポーシスを表現する様々な言葉が使われてきた:FAP,軽症型FAP,Gardener 症候群,Turcot症候群,胃腺がんおよび胃近位ポリポーシス(GAPPS).これらの表現型に関する臨床的特徴はこのGeneReviewに記されている;しかしながら,現在ではこれらすべてがAPC遺伝子の病的バリアントに起因することが遺伝学的に定義されている.Gardener 症候群およびTurcot症候群は,現在はFAPの一部として知られているため,過去のものとして現在は使用するべきではない.
頻度
FAPの頻度は,6,850~31,250人に1人(人口10万人あたり2.29-3.2人)と幅がある [Half et al 2009, Scheuner et al 2010].世界中で発症頻度の地域差はなく,男女差もない.軽症型FAPは,FAPに比べてポリープ数が少なく大腸がんのリスクも低いため,診断されていない可能性がある [Neklason et al 2008].
GAPPSの頻度は,今のところわかっていない.
APC関連ポリポーシスは,かつて全大腸がんの0.5%を占めると考えられてきた;リスクのある家族に対して早期のポリープ発見と予防的大腸切除術が行われるようになったため,この数字は低下しつつある.
遺伝的に関連がある疾患
APC遺伝子の生殖細胞系列病的バリアントに関連することが知られている表現型は,このGeneReviewに記されているもの以外はない.
5q22の欠失.APC遺伝子を含む5q22の染色体中間部欠失が,軽症型腺腫性ポリポーシス[Pilarski et al 1999]および古典型腺腫性ポリポーシス [Heald et al 2007]の患者で報告されている.どの報告においても,これらの患者は認知機能障害があり,その症状は通常軽度~中等度で,大半は身体の形態異常を有する [Heald et al 2007].
APC関連ポリポーシスの他の所見のない,散発性腫瘍(大腸含む)はAPC遺伝子の体細胞病的バリアントから発生するが,生殖細胞系列にはこのバリアントはない.この状況においては,この腫瘍に遺伝性はない.より詳細な情報は,がんと良性腫瘍の項を参照.
鑑別診断
APC関連ポリポーシスは他の大腸がんを伴う遺伝性疾患や他の消化管ポリポーシス症候群(Table 4参照)とは分子遺伝学的検査,病理学的所見や臨床的特徴によって鑑別される.
表4.APC関連ポリポーシスの鑑別診断にあがる遺伝性ポリポーシスおよび大腸がん症候群
| 遺伝子/遺伝的機構 | 疾患 | MOI | 備考 |
|---|---|---|---|
| 15q15.3q22.1 重複1 BMPR1A SMAD4 |
遺伝性混合ポリポーシス症候群(Hereditary mixed polyposis syndrome, HMPS)(OMIM 601228) | AD | 大腸がんのリスク上昇および様々な異なる種類の大腸ポリープと関連する.特徴的な病変は,若年性腺腫性大腸ポリープの混合型である.腺腫,過形成鋸歯状腺腫,混合型過形成腺腫性ポリープも発生する. |
| AXIN2 | AXIN2-関連ポリポーシス (減歯症-大腸がん症候群) (OMIM 608615) | AD | 外胚葉形成不全 |
| BMPR1A SMAD4 |
若年性ポリポーシス症候群 (JPS) | AD | FAPとJPSを鑑別する所見としてよく挙げられる,過誤腫性ポリープの易罹患性を特徴とする.過誤腫性ポリープは消化管-特に胃,小腸,結腸,直腸に発生する.JPSの多くの患者は20歳までにポリープを発症する.ほとんどの若年性ポリープは良性だが,悪性化するとがんを発症しうる. |
| EPCAM MLH1 MSH2 MSH6 PMS2 |
リンチ症候群 (遺伝性非ポリポーシス大腸がん) | AD | 若年発症の大腸がんで数個の腺腫性大腸ポリープがある場合,軽症型FAPとリンチ症候群を鑑別するのは難しい場合がある.大腸以外のがんの家族歴や所見,腫瘍組織のMSI検査やIHC検査が鑑別に役立つかもしれない. |
| MLH1 MSH2 MSH6 PMS2 |
先天性ミスマッチ修復欠損 (CMMRD) (リンチ症候群参照.) | AR | 罹患者は幼少期より脳腫瘍,血液悪性腫瘍,大腸がんやその他のリンチ症候群関連腫瘍を発症する.カフェ・オ・レ班や腋窩/鼠径部の小斑点もほとんどの患者にみられる;軽症型FAPに似た複数の大腸腺腫も認められる. |
| MSH3 | MSH3-関連ポリポーシス (OMIM 617100) | AR | 大腸と十二指腸腺腫,大腸がん,胃癌,若年発症の星細胞腫 |
| MUTYH | MUTYH ポリポーシス (MAP) | AR | 多数の腺腫および大腸ポリポーシスの易罹患性と関連する.MAPの大腸の表現型は軽症型FAPと類似する.大腸ポリポーシスがあってAPCに病的バリアントが認められない場合,MUTYHの分子遺伝学的検査を検討するべきである. |
| NF1 | 神経線維腫症1型 (NF1) | AD | NF1患者では,小腸・胃・大腸に複数の消化管ポリープ状神経線維腫や神経節細胞腫を生じうる. |
| NTHL1 | NTHL1 腫瘍症候群 (NTHL1-関連ポリポーシス) | AR | 大腸がん,乳がん,大腸ポリポーシスの生涯リスクが高まる.その他のがん種では,子宮体がん,子宮頸がん,膀胱尿路上皮がん,髄膜腫,不特定の脳腫瘍,基底細胞がん,頭頸部扁平上皮がん,血液悪性腫瘍も含まれる. |
| POLD1 | POLD1-関連ポリポーシス (ポリメラーゼ校正-関連ポリポーシス) (OMIM 612591) | AD | 大腸と十二指腸の腺腫;MDPL症候群(下顎骨低形成,難聴,早老症,脂肪異栄養症) |
| POLE | POLE-関連ポリポーシス (ポリメラーゼ校正-関連ポリポーシス) (OMIM 615083) | AD | 大腸と十二指腸の腺腫 |
| PTEN | カウデン症候群 (CS) (See PTEN 過誤腫症候群.) | AD | CSは複数の大腸ポリープと関連するが,(APC関連ポリポーシスの病態とは異なり) 過誤腫性ポリープや若年性ポリープ,脂肪腫,神経節腫が多い.CSでは大腸がんのリスクが上昇するが,乳がん・甲状腺がん・子宮体がんも一般的である. |
| STK11 | Peutz-Jeghers症候群 (PJS) | AD | 消化管のPJS型ポリープと粘膜上皮の色素沈着を特徴とし,これらはAPC関連ポリポーシスではみられない.PJS ポリープは有症状であることが多く,小腸で頻発する(空腸,回腸,十二指腸の順に多い)が,その他の消化管のどこでも発生しうる. |
AD=常染色体優性;AR=常染色体劣性;FAP=家族性大腸ポリポーシス;IHC=免疫組織化学;MOI=遺伝形式;MSI=マイクロサテライト不安定性
- HMPSはBMPR1Aの病的バリアントまたはGREM1の過剰発現を引き起こす15q15.3q22.1の重複のどちらでも生じうる[Jaeger et al 2012].遺伝性混合ポリポーシス症候群ではSMAD4の病的バリアントを持つ家系も存在する[Valle et al 2019].
鑑別診断を考慮すべき後天的疾患
Cronkite-Canada症候群は,消化管全般の過誤腫性ポリポーシス,皮膚の過剰色素沈着,脱毛,爪の萎縮を特徴とする.
結節性リンパ性過形成は,小腸,胃,結腸にリンパ節の過形成をきたすリンパ性増殖性疾患で,分類不能型免疫不全症(CVID)との関連があると考えられている.
リンパ腫様ポリポーシスは,消化管における原発性節外性リンパ腫の発生を特徴とする.多発性リンパ腫様ポリポーシスと地中海型リンパ腫の2病型がある.
炎症性ポリポーシスは,炎症性腸疾患に伴う後天性の非腫瘍性ポリープを特徴とする.
散発性大腸腫瘍.大部分の家族性ではない大腸腫瘍では,APC遺伝子の体細胞病的バリアントが関連している(遺伝的に関連のある疾患の項,参照).[Lüchtenborg et al 2004, Christie et al 2013] この変化は大腸腫瘍の発生初期に起こると考えられている[Christie et al 2013, Aghabozorgi et al 2019].
治療関連ポリポーシスは,消化管ポリポーシスの原因として報告されている [Biller et al 2020].8施設34人の消化管ポリポーシス患者で,ポリポーシス所見の少なくとも10年以上前に幼少期または若年がんの治療として放射線治療および/または化学療法を受けていた [Biller et al 2020].しかし,消化管ポリポーシスが化学療法/放射線治療によるものなのか,これらの患者にたまたま偶発的に起きたものなのかは不明である.
その他
原因不明の大腸腺腫性ポリポーシス.大腸腺腫性ポリープ(累積大腸ポリープ10個以上)のある多くの患者では,生殖細胞系列分子遺伝学的検査で情報が得られない可能性が高い[Stanich et al 2019].注:これらの患者では,未検出のAPC体細胞モザイクの可能性を考慮するべきである[Jansen & Goel 2020].
鋸歯状ポリポーシス症候群(以前の名称は過形成ポリポーシス)は,多発性大腸鋸歯状ポリープ(過形成ポリープ,sessile serrated adenomas/polyps [SSA/P] および古典型鋸歯状腺腫)を含む.本症は遺伝性かそれとも後天的なのか不明である [Snover et al 2010];RNF43の病的バリアントが,ごく一部の患者に関連している可能性がある[Yan et al 2017].通常,鋸歯状ポリープが優性であるが,鋸歯状ポリポーシスの患者は多発性大腸腺腫を合併することも多い [Kalady et al 2011].大腸がんの家族歴が見られる場合もあるが,鋸歯状ポリポーシス症候群の診断基準を満たす家族が複数いることは稀である.
臨床的マネジメント
初期診断後の評価
古典型家族性大腸ポリポーシス(FAP)または軽症型FAPと診断された患者は,この節で概要を述べるように,症状の治療だけでなく,サーベイランスおよび一次予防のための年齢に応じた推奨事項について,カウンセリングを受けるべきである.現段階では,胃腺がんおよび胃近位ポリポーシス(GAPPS)に対する合意のとれた管理ガイドラインはない.
症状に対する治療
手術に関する情報を含む診療パラメーターについて,以下の専門家グループによって概説されている.
- 米国総合癌ネットワーク(NCCN) [Weiss et al 2021] (全文)
- 米国消化器内視鏡学会 [Yang et al 2020] (全文)
- 米国消化器病学会 [Syngal et al 2015] (全文)
- 米国大腸外科医学会 [Herzig et al 2017] (全文)
- 米国臨床腫瘍学会 [Stoffel et al 2015] (全文)
- 英国消化器病学会 [Monahan et al 2020] (全文)
- 外科腫瘍学会[Guillem et al 2006] (全文)
- デスモイド腫瘍研究会 [2020] (全文)
- 欧州消化器内視鏡学会[van Leerdam et al 2019] (全文)
大腸ポリープ.FAP患者に対しては,大腸内視鏡による内視鏡的サーベイランスを,1~2年ごとに10~15歳から開始することが通常推奨される[Weiss et al 2016, van Leerdam et al 2019, Yang et al 2020].5mmを超えるポリープはすべて切除するべきである.ポリープの数が管理可能で手術の決定的な適応がない場合は,大腸切除を遅らせ,内視鏡的サーベイランスを行うことが適切である [Ishikawa et al 2016].大腸切除術の絶対的適応は,大腸がんと報告されたあるいは疑わしい状態,もしくは重大な症状(腸閉塞や下血)が表れた場合である.これらは,がんが存在しなければめったに起こらない.大腸切除術の相対的適応は,内視鏡検査で管理しきれない複数の腺腫(>10㎜)がある場合や,経過観察中に腺腫の数が顕著に増加する場合,高度異形成の腺腫がある,もしくは大腸検査が適切に実施できない(例えば,無数の小型腺腫の存在や大腸内視鏡検査のアクセスや追従が制限される)場合などが含まれる.10~20歳の患者で,腺腫が6mm未満かつ絨毛性変化や高度異形成がなければ,身体的・精神的な成熟を考慮し、大腸切除の時期を遅らせることも考えられる.
軽症型FAP患者の表現型では,大腸内視鏡検査の開始を青年期後半以降に遅らせることも可能である[Weiss et al 2016, van Leerdam et al 2019, Yang et al 2020].1~2年ごとの大腸内視鏡によるサーベイランスは有効であることが多く,大腸切除術を遅らせることやその必要性がなくなることさえある[Knudsen et al 2010].約1/3の患者では,ポリープの数が少ないため,定期的なポリープ切除を伴う大腸内視鏡サーベイランスで十分ある( サーベイランスの項,参照) [Patel et al 2016].
大腸切除の術式を以下に示す:
- 大腸全摘・回腸囊肛門(管)吻合術(IPAA) .腹腔鏡下,補助的な腹腔鏡下,または開腹で実施される.IPAAは,肛門移行上皮および低位直腸粘膜1~2cmを残してステープルで留める,あるいは肛門粘膜を完全に切除した後,手で縫合する.これは多段階手術となる.
- 結腸全摘・回腸直腸吻合術(IRA).最小限の侵襲的手術手技で実施可能で,単回手術である.
- 大腸全摘・回腸人工肛門造設術.最小限の侵襲的手術手技で実施可能で,単回手術である.
手術の選択は、臨床的状況による。
- IPAAは,FAP患者では通常,直腸のポリープによる症状が重い(一般的には,20個を超える直腸の腺腫または進行した直腸の異形成)あるいは,直腸の病態が内視鏡的に管理できなくなった時点でIRAの次の処置として実施される [Warrier & Kalady 2012].この処置の長所は,直腸がんのリスクをほぼ排除し,腸の機能を比較的いい状態で温存できることである.ただし,IRAによる大腸切除術と比較して,膀胱/性機能不全のリスクが高まる可能性があり,機能的帰結は様々である.
FAP患者と回腸嚢に関する研究では,57%の回腸嚢に腺腫性ポリープが見つかった.[Groves et al 2005].
- IRAは一般的に,直腸のポリープによる症状が少なく,内視鏡的管理が可能と考えられる場合に検討される(通常,軽症型FAPの場合に用いられる).合併症率が低く,技術的に簡単な処置である.通常,機能的帰結は良好であり,性または尿路機能不全のリスクは最小限である.適切な対象者に実施すれば,直腸がんのリスクやIRA後に全大腸切除術が必要となる可能性は低い[Church et al 2001].この処置は,重症の直腸疾患があったり,術後に残存直腸の内視鏡によるサーベイランスを確実に受けることができない患者に対しては実施すべきではない.
- 大腸全摘・回腸人工肛門造設術は,(直腸ポリープ/がんによる症状が重いために)大腸全摘術が必要となったり,IPAAへの禁忌(回腸嚢を骨盤底につなげるのを妨げるような腸間膜デスモイド,骨盤底に浸潤する低位直腸がん,あるいは括約筋の制御不良がある患者の希望など)があるという場合でない限り,めったに必要となることはない.
十二指腸腺腫.最近のガイドラインでは, 20~25歳あるいは大腸切除術を予定する場合はそれよりも前に,十二指腸のスクリーニングを行うことを推奨している[van Leerdam et al 2019, Yang et al 2020, Weiss et al 2021].この推奨は,大腸の表現型によらず,古典型,軽症型FAPともに適用される.サーベイランスの間隔はSpigelman分類に基づいて決定される [Spigelman et al 1989].この分類は,十二指腸ポリープの数,大きさ,組織像,異形度に応じて5段階に分類される(Table 5参照).ガイドラインは概ね低リスク患者(Stage0・Ⅰ)に対しては5年ごとの十二指腸スクリーニングを,中程度リスク(StageⅡ)の患者では3年ごとを推奨している.高リスク患者(StageⅢ・Ⅳ)では少なくとも1年ごとの内視鏡検査が必要とされる;StageⅣの患者では,外科に紹介することも検討されるべきである.
表5.家族性大腸ポリポーシスの十二指腸腺腫に対するSpigelman分類
| Spigelman 分類 | |||
|---|---|---|---|
| 基準 | 1点 | 2点 | 3点 |
| ポリープ数 | 1-4 | 5-20 | >20 |
| 最大径(mm) | 1-4 | 5-10 | >10 |
| 組織像 | 管状 | 管状絨毛 | 絨毛 |
| 異形度 | 低 | 中 | 高 |
Stage 0 = 0点
Stage I = 1-4点
Stage II = 5-6点
Stage III = 7-8点
Stage IV = 9-12点
十二指腸および/または膨大部腺腫の内視鏡的または外科的切除は,スネアによる摘除や内視鏡的粘膜切除術などの標準的なポリープ摘除術によって行うことが推奨される.摘除しきれない数のポリープがある場合は,1cmを超える大きさまたは気になる特徴のあるポリープに焦点をあてて摘除する.欧州消化器内視鏡学会のガイドライン[van Leerdam et al 2019]では,小さいポリープの摘除については,線維化して将来的な切除の制限となる懸念があり注意が必要とされているが,臨床の現場では,これが制限要因になるという報告はない.
進行した十二指腸ポリポーシスに対する手術適応となるのは,高度異形成を伴うStage III,Stage IV,悪性病変である.手術の選択肢には,膵頭十二指腸切除術(Whipple法) や膵温存十二指腸切除術があり,後者は乳頭部に腺腫がなく,がんの疑いがない場合に有効な選択肢となる.これらの手術は合併症のリスクが高いため,手術実績の多い病院で,できればFAPの専門家によって行われるべきである.
膨大部腺腫.十分な検査を行うためには,特殊な内視鏡技術が必要である.これが可能となるのは,側視十二指腸内視鏡と直視胃内視鏡の先端に透明なキャップを装着する方法があり,この2つの方法の成果は同等である [Abdelhafez et al 2019].膨大部の生検は,膵炎のリスクは低く安全であることが示されており,腺腫の疑いがあれば生検を行う閾値は低くあるべきである[Mehta et al 2021].膨大部の腺腫を,サーベイランスの間隔を決定するための十二指腸ポリポーシスに含めるかどうかは,ガイドラインによって様々である;膨大部に小さな腺腫を見つけた場合,多くの専門家は3年ごとのサーベイランスを推奨している.小さな膨大部の腺腫は摘除せずに経過観察が可能であるが,1cmを超える大きさの腺腫や進行した組織像を呈するものは摘除するべきである.膨大部切除術は合併症を起こすことが多く,経験豊富な医師によって行われることが望ましい[Roos et al 2021].膨大部切除後に再発することも一般的であるため,頻回な内視鏡的サーベイランスが必要とされる.膨大部の腺腫に対する手術の検討は,進行した十二指腸ポリポーシスと同様である.
胃のポリープ.胃のサーベイランスは,十二指腸のサーベイランスと同時に行う.胃のサーベイランスについては,十二指腸ポリポーシスに対するものほどガイドライン上の推奨は確定していないが,FAP患者の胃がん発症が増えてきているので,これらのガイドラインも進化していくものと思われる.腺腫/幽門腺腺腫や進行形の変化(異形成)が懸念されるポリープは,胃底腺ポリープ(FGPs)のランダムサンプリングとともに,すべて切除することが推奨される.専門家によっては,ポリープの数,大きさ,組織像,異形度,その他の特徴などをサーベイランスの方針に役立てることを推奨している[Mankaney et al 2017];内視鏡の基準(内視鏡検査におけるがんに関連する病態のサーベイランス; Mankaney et al [2020]参照)によって,リスクの高い病理像の視覚的診断に役立てることができる.
甲状腺結節およびがん.甲状腺結節および櫛型を含む甲状腺乳頭がんの治療は,散発性の疾患と同様である [Abdullah Suhaimi et al 2015].
骨腫は,整容的理由により切除することがある.
デスモイド腫瘍.可能な治療法としては,外科的切除(頻回に再発する場合),非ステロイド性抗炎症薬(NSAIDs),抗エストロゲン薬,細胞障害型化学療法,放射線照射などがある[Smith et al 2000a, Tonelli et al 2003, Gega et al 2006].デスモイド腫瘍に対する治療法の総説は,Guillem et al [2006]とDesmoid Tumor Working Group [2020]で閲覧できる.
副腎腫瘍.副腎腫瘍に対しては,必要に応じて標準的な治療が適応となる.
化学的予防.現時点では,FDAに承認されたFAPに対する化学予防薬はない.化学予防に関心のある患者は,現在進行中の臨床試験への参加を勧める(研究中の治療の項参照).注:FDAの見解としては,腺腫の数や大きさの変化が,承認するには不十分であり,臨床的有用性の明確な科学的根拠が必要であるとしている.臨床的有用性の例として,大腸がんのリスク低減や手術の必要性の削減が挙げられており,現在の臨床試験はこれらを最終評価項目として設計されている.
NSAIDs.スリンダク(sulindac)の非プラセボ対照試験および観察試験は,ポリープの大きさと数を顕著に減少させ,初めのうちは有望だった.しかしながら,これらの予備研究はそのデザインに限界があった(非プラセボ対照試験であり,患者数が少なく,直腸のみ検査可能という患者も含まれていた).その後,いくつかの対照試験において,スリンダクによる治療中にポリープによる症状を軽減したことが確認された [Labayle et al 1991, Giardiello et al 1993a, Nugent et al 1993].しかしながら,スリンダク中止後,急速な再発またはポリープ数の増加が見られた[Labayle et al 1991, Giardiello et al 1993a].その後の,APC遺伝子に病的バリアントのある人のポリープの一次予防を評価するためにデザインされた研究では,プラセボ群と比較して統計的有意ではないものの,有効性の傾向が示された[Giardiello et al 2002].
FDAは当初,結腸におけるポリープによる症状や大きさを軽減させる(同様に十二指腸でも軽減させる)という科学的根拠に基づき,FAP患者に対してセレコキシブ(celecoxib)の処方を承認した[Steinbach et al 2000, Phillips et al 2002].しかし,心血管および脳血管への安全性に懸念があるとして,FAP患者に対するセレコキシブのFDA承認は取り下げられ,ロフェコキシブ(rofecoxib)は市場から完全に撤退した.
アスピリンは従来,FAPではほとんどあるいは全く有益でないことが示されていた [Burn et al 2001, Ishikawa et al 2013]が,最近の無作為化比較試験で大きいポリープの抑制効果の可能性が示された[Ishikawa et al 2021].
スリンダクとジフルオロメチルオルニチン(DFMO)の併用により,散発性の異時性腺腫が顕著な減少を示したという報告があり,NSAIDsと他の薬剤との組み合わせへの関心が高まった[Meyskens et al 2008].92名のFAP患者が参加した無作為化プラセボ対照試験では,スリンダクとエルロチニブ(EGF受容体阻害剤)の6か月間の併用により,プラセボと比べ,十二指腸ポリープの症状が軽減した[Samadder et al 2016].有害事象は,投薬群に共通して見られた(87%にニキビ様発疹)が,重大な有害事象はほとんどなかった(全参加者のうち2名) [Samadder et al 2016].この試験の二次解析では,大腸ポリープの症状軽減も示された[Samadder et al 2018].この試験に対しては,最終評価項目が明確に臨床的な意味を持たないことが批判された.セレコキシブ単剤とセレコキシブ+DFMOの併用を比較したところ,定義した内視鏡視野内でのポリープの症状の明らかな違いは見られなかった(しかし,より広範囲の映像評価を使用した場合,薬剤を併用した治療群でポリープの症状軽減が見られた) [Lynch et al 2016].最近では,大きな臨床試験で,スリンダクとエフロルニチン(eflornithine)の併用およびそれぞれの単剤使用で,疾患の進行に違いは見られなかったと報告された[Burke et al 2020].
注:大腸切除術前のNSAIDsの使用はまだ試験段階にある(研究中の治療の項参照).
GAPPS.GAPPSに対する疾患管理ガイドラインは今のところ存在しないが,専門家によっては,15歳から胃内視鏡を開始し,腺腫/幽門腺腺腫や進行形の変化(異形成)が懸念されるポリープは,FGPsのランダムサンプリングとともに,すべて切除することを推奨している.抽出した中に進行した腫瘍が見つかれば,標準的な外科的胃切除術を考慮するべきである[筆者の私見].
一次予防
FAP.古典型FAP患者の大腸がんリスク低減のために,大腸切除術が提案される.軽症型FAPの患者に対しては,大腸切除術が必要となることもあるが,約1/3の患者では,大腸ポリープの数が少ないため,定期的な大腸内視鏡下ポリープ切除術によっても,十分大腸がんを予防可能である.
GAPPS.GAPPS患者に対して予防的胃切除術をするべきかどうかは,現在のところはっきりしていない.専門家によっては,おおよそ30歳くらいで予防的胃切除術を考慮することを推奨している[Tacheci et al 2021].
サーベイランス
複数の専門家団体が,現時点でのエビデンスに基づき,専門家のコンセンサスもふまえてガイドラインを公表している[Syngal et al 2015, Herzig et al 2017, Weiss et al 2021].以下のサーベイランスの推奨事項は,これらの専門家団体のガイドラインに基づいている.
表6. FAP患者に対して推奨されるサーベイランス
| 臓器/関連事項 | 評価項目 | 頻度/備考 |
|---|---|---|
| 大腸腺腫性ポリープ | 大腸内視鏡検査 | 1-2年ごと 開始時期:古典型FAPでは10-15歳;軽症型FAPでは青年期後半 |
| IPAAによる大腸全摘術を受けた人: 回腸嚢の内視鏡的サーベイランス | 1-2年ごと | |
| 結腸全摘・回腸直腸吻合術を受けた人: 残存直腸に対するサーベイランス |
|
|
| 大腸全摘・回腸人工肛門造設術を受けた人: 回腸内視鏡 | 1-2年ごと | |
| 小腸ポリープ&がん | Vater膨大部を完全に可視化したEGD (十二指腸鏡または透明なキャップを用いる) | 6ヶ月-5年ごと 十二指腸腺腫の症状による2 開始時期:20-25歳または大腸切除術の前 |
| 特に(Spigelman分類に基づいて)十二指腸ポリポーシスが進行している場合は,小腸を完全可視化するカプセル内視鏡またはCT/MRエンテログラフィを考慮する. | ||
| 甲状腺がん |
|
2-5年ごと 開始時期:青年期後半3 |
| CNS 腫瘍 | 神経学的検査 | 診断されたら1年ごと |
| 肝芽腫 |
|
3-6ヶ月ごと 生後5歳まで4 |
| 胃のポリープ&がん | EGD |
|
| 非腫瘍性大腸外病変 | 大腸外病変に対する検診 (骨腫,歯牙異常,皮膚病変など) | 1年ごと |
| デスモイド腫瘍 | 腹部の触診 | 1年ごと |
| MRI またはCT スキャン | 定期的なスクリーニングは推奨されていない; しかしながら,大腸切除術後,説明できない症状が出現した場合は,隣接する臓器を圧迫する可能性のあるデスモイドに対する検査を行う閾値は低くあるべきである.5 | |
| 副腎腫瘍 | 根拠のあるスクリーニング法はない |
CNS=中枢神経系;EGD=食道・胃・十二指腸内視鏡;FAP=家族性大腸ポリポーシス;IPAA=回腸囊肛門(管)吻合術
- Church et al [2003]
- EGDの頻度は十二指腸腺腫の重症度による;Spigelman分類が頻度を決定する一助となる.Spigelman分類は Syngal et al [2015]によってまとめられた.Table 5も参照のこと.
- Syngal et al [2015], Herzig et al [2017], Weiss et al [2021]
- Weiss et al [2021]
- デスモイド腫瘍のスクリーニングについてのデータは限定的である.
GAPPS.現時点ではGAPPS患者に対して胃がんのスクリーニングをすべきかどうかははっきりしていない.胃ポリポーシスの程度にもより,FGPsが急速に進行するという報告もあるため,胃がんのサーベイランスの効果は限定的である [Repak et al 2016].
避けるべき薬剤や環境
手術とデスモイドのリスク.腹部手術によってデスモイド腫瘍のリスクが増加することは証明されており,2段階の手術を必要とする外科的処置においてより高まる.デスモイドのリスクが高い患者(女性,APC遺伝子のコドン1395-1493の病的バリアント,デスモイドの家族歴など)では,再手術が必要となる可能性を最小限に留めるために,単回手術で済む可能性が高い術式を検討するべきである.
手術と生殖能力.女性では,IPAAによる大腸全摘後に,生殖能力が低下する可能性がある [Rajaratnam et al 2011].ほとんどが炎症性腸疾患に対する手術の結果から得られた情報ではあるが,女性のFAP患者では術式の選択の際に,議論に含める必要がある[Olsen et al 2003].
リスクのある血縁者に対する検査
罹患者の第一度近親者(両親,同胞,子)について,APC病的バリアントの分子遺伝学的検査を行って遺伝学的状態を明らかにすることは適切である.リスクのある血縁者に対して早期に同定するために分子遺伝学的検査を行うことは,診断をより正確なものとし,病的バリアントを受け継いでいない家系員に対して侵襲的なスクリーニング( サーベイランスの項参照)を減らすことができる.
- 早期にAPC関連ポリポーシスを認識することで,タイミングよく介入を始められ,最終的な帰結を改善できる可能性がある.
- 罹患した血縁者の情報からAPC関連ポリポーシスと診断された人は,症状に基づいて診断された人に比べ,圧倒的に寿命が長い.
FAPのリスクのある人に対する大腸スクリーニングは早ければ10-12歳から始まるので,分子遺伝学的検査は一般に10歳までに提供される.親や小児科医は,罹患児に対して乳児期から5歳までの肝芽腫のスクリーニングを考慮するので,出生時の遺伝学的検査も必要とされる場合がある.軽症型FAPの場合,大腸スクリーニングは青年期後半から開始されるので,分子遺伝学的検査もそれまで遅らせてもよい.親は,病的バリアントを受け継いでいない子に不必要な処置を受けさせないために,スクリーニング開始前に子の遺伝学的状態を確認したがることが多い.
注:スクリーニングを開始する至適年齢についての確証はない.したがって何歳でテストを実施し,スクリーニングを開始するかは医療機関,家族歴,肝芽腫のスクリーニング,親や子の希望によって変わってくる.
妊娠の管理
妊娠/生殖/ホルモン剤の使用.FAP患者の女性が妊娠することへの影響があるのかどうかという情報は限定的である.162名のFAP女性の妊娠率を,2つの大腸術式の前後とコントロール群で比較していた研究がある.手術を実施していないFAP女性ではコントロール群の一般女性と同様に妊娠した.さらに, IRAの術式を選択したFAP女性もコントロール群と同様の妊娠率を示した. IPAAによる大腸全摘術を実施したFAP女性では,コントロール群と比べ,著しい妊娠率の減少が見られた[Olsen et al 2003].
別の研究では,IRA,IPAAおよび大腸全摘・回腸人工肛門造設術を実施したいずれのFAP患者においても,自己申告による妊娠の問題に違いは見られなかった.しかしながら,最初の手術をより若年で実施した者は,術後に妊娠の問題が発生する頻度が高まった[Nieuwenhuis et al 2010].
デスモイド腫瘍の発生や進行と妊娠との関係性については,支持する根拠は限定的で[Sinha et al 2011],妊娠後のデスモイドの経過がより良好であるという報告がある[Church & McGannon 2000].
大腸摘出術を行った女性の産科関連合併症のリスクは,その他の主要な腹部手術をした女性と変わらないと考えられ,こういった手術をしていない女性と比べ,帝王切開による出産が多い.
大腸切除術を実施した時点のFAP女性に関する研究では,妊娠歴と大腸ポリープの重症度には関連は見られなかったが,重症の十二指腸疾患をもつ割合は,経産女性の方が未経産女性に比べて明らかに高かった[Suraweera et al 2007].
いくつかの研究では,一般集団においては,女性ホルモンは大腸がんの発生に予防的に働くことが示唆されている.ある女性では,経口避妊薬を使用した後,ポリープが減少した[Giardiello et al 2005].
研究中の治療
単一のコントロール群をおいた試験では,ω3多価不飽和脂肪酸であるエイコサペンタエン酸(EPA)はFAP患者のポリープの大きさと数を20-30%減少させた [West et al 2010].IRAによる結腸全摘術後の直腸ポリープの症状に対するEPAの効果をみる,phaseⅢ多施設共同試験が現在実施されている(NCT03806426).オベチコール酸,lorpucitinib(汎JAK阻害剤),カプセル化シロリムス,およびグセルクマブ(IL-23阻害剤)についても,多くの多施設共同試験が実施されている.
広範な疾患や症状の臨床研究に関する情報は,米国については ClinicalTrials.govを,ヨーロッパについてはEU Clinical Trials Registerを参照すること.
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
APC関連ポリポーシス-家族性大腸ポリポーシス(FAP),軽症型FAP,胃腺がんおよび胃近位ポリポーシス(GAPPS)-は常染色体顕性遺伝(優性遺伝)の形式をとる.
患者家族のリスク
発端者の両親
- APC関連ポリポーシスと診断された患者の大部分には,罹患した親がいる.
- APC関連ポリポーシスと診断された患者のうち,およそ25%はde novo (新たな)病的バリアントの結果,発症する[Aretz et al 2004].
- 発端者が家族の中で1人だけ罹患している(つまり孤発例)場合,分子遺伝学的検査は発端者の両親に勧められる.
- 発端者で同定された病的バリアントが両親で見つからず,両親が生物学的に母と父であることが確認できている場合は,以下の可能性を考慮する
- 発端者がde novo (新たな)病的バリアントを保持している.
- どちらかの親の生殖細胞系列(または体細胞と生殖細胞系列)モザイクで,発端者が病的バリアントを受け継いだ.* 生殖細胞系列モザイクは多数の家系から報告されている[Hes et al 2008, Schwab et al 2008, Spier et al 2016].注:親の白血球DNAの検査では,体細胞モザイクは全例検出できない可能性があり,生殖細胞系列のみに存在する病的バリアントは検出できない.
* APC遺伝子の体細胞と生殖細胞系列モザイクを持つ親は,軽度/最小限の影響を受けている可能性がある.ある研究では,APC遺伝子の体細胞モザイクの患者の多く(65%)は,20~100個の腺腫がみられ(軽症型FAP),30%はFAPの表現型を呈し,5%は腺腫がみられなかった[Jansen & Goel 2020].
- 家族がこの疾病を認識していなかったり,親が発症前に早期に死亡していたり,あるいは罹患した親の発症年齢が高かったりするために,APC関連ポリポーシスと診断された患者で家族歴が明らかでない場合もある.そのため,発端者で同定されたAPC病的バリアントについて,両方の親に対して分子遺伝学的検査が行われていない限り、明らかに陰性という家族歴は確認できない.
発端者の同胞
発端者の同胞のリスクは両親の遺伝的状況による.
- 両親のいずれかが罹患者かAPC遺伝子に病的バリアントを有している場合は,同胞が病的バリアントを受け継ぐリスクは50%である.ヘテロ接合体の家族内では表現型にばらつきが見られる(浸透率 と臨床像の項参照).APC病的バリアントを受け継いだ同胞に対する推奨事項は,サーベイランスと 一次予防の項を参照のこと.
- 両親の白血球DNAのいずれにも発端者に認められたAPC病的バリアントが同定されなかった場合,親のモザイクの可能性があるため,一般集団よりはわずかに高い.したがって,明らかにde novo (新たな)病的バリアントと思われる場合であっても同胞の分子遺伝学的検査が考慮されるべきである.
- 生殖細胞系列モザイクは無症状の79歳の女性で確認されている.彼女の2人の息子は数千個の腺腫性大腸ポリープとAPC病的バリアントを有している[Hes et al 2008].
- 他にも,母親が無症状の生殖細胞系列モザイクで,2人の子どもに大腸腺腫性ポリポーシスと,結果的にAPC病的バリアントが同定された例も報告されている[Schwab et al 2008].
発端者の子
- FAP患者の子はそれぞれ,50%の確率でAPCの病的バリアントを受け継ぐ.
他の血縁者
他の血縁者のリスクは発端者の両親の状況による.両親のいずれかがAPC病的バリアントを有している場合は,その親の血縁者にもリスクがある.
遺伝カウンセリングに関連した問題
症前検査(リスクがあり無症状の者に対する検査)
- 罹患した家系員でAPC遺伝子に病的バリアントが同定されれば,リスクのある血縁者の発症前検査は可能である.
- こういった検査の結果として考えられることを-社会経済的変化に限らず,陽性結果だった場合の長期間のフォローアップや検査体制の必要性なども含め-発症前検査の特性や限界と同様に,検査前のきちんとした遺伝カウンセリングの場で議論されなければならない.
- リスクのある若い家系員に対する発症前の分子遺伝学的検査を検討することは,医学的管理に導くために適切である(疾患の管理および 患者家族のリスクの項参照).遺伝学的検査の前には子と両親に対する教育に関して特別の注意を払う必要がある.検査結果を両親と子に伝える方法についてはあらかじめ決めておくべきである.大部分の子どもは発症前遺伝学的検査結果の開示後に臨床的に重要な心理的問題を生じてはいないが,Hyer et al [2019]はこうした家族に対する長期の心理的サポートを準備することを推奨している.
臨床的に診断された血縁者の検査情報が入手できない状況で,リスクのある家族の遺伝的状態を判断するために分子遺伝学的検査を行うのは問題を含んでおり,またその結果の解釈には注意を要する.家族での結果が陽性であった場合は,その家系内にAPC遺伝子の病的バリアントが存在することを意味し,同じ検査法が家系内の他のリスクのある者の,遺伝的状態の評価に利用できることを示す.一方,罹患している患者よりも先に家族に対して分子遺伝学的検査が行われる場合,病的バリアントが見つからなかったからといって,他の家族にAPC遺伝子の病的バリアントが存在する可能性がなくなるわけではない.
遺伝性腫瘍のリスク評価とカウンセリング.
分子遺伝学的検査の実施の如何に関わらず,がんのリスク評価の過程でリスクのある者を同定することの,医学的,心理学的,倫理的な包括的説明はCancer Genetics Risk Assessment and Counseling - for health professionals (part of PDQ®,米国国立がん研究所)を参照すること.
家族計画
- 遺伝学的リスクを判定および出生前/着床前診断を利用するかどうかの議論は,妊娠前に行うのが望ましい.
- 罹患している,もしくはリスクのある若年成人に対しては,遺伝カウンセリング(子どもがリスクを持つ可能性や生殖医療における選択肢に関する検討を含む)を提供するのが適切である.
出生前診断および着床前の遺伝的診断
APC遺伝子の病的バリアントが罹患者で同定されれば,リスクの高い妊娠のための出生前診断や着床前の遺伝学的診断が可能である.胎児においてAPC病的バリアントを検出しても発症時期や重症度の予測はできないことに注意する必要がある[Rechitsky et al 2002, Davis et al 2006].
医療の専門家間や家族内においても,出生前診断に対する考え方の相違が存在しうる.多くの専門機関は出生前診断については夫婦の自己決定の問題だと考えているが,この問題については議論することが適切である.
訳注:日本では本症における出生前診断および着床前診断は行われていない.
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- 大腸ポリポーシス患者友の会「ハーモニー・ライン」
- 大腸腺腫症患者,家族および協賛者の会「ハーモニー・ライフ」
- Collaborative Group of the Americas on Inherited Gastrointestinal Cancer (CGA-IGC)
- MedlinePlus
- National Cancer Institute (NCI)
6116 Executive Boulevard
Suite 300
Bethesda MD 20892-8322
Phone: 800-422-6237 (toll-free)
Email: cancergovstaff@mail.nih.gov
Genetics of Colorectal Cancer (PDQ®)
- American Cancer Society
Phone: 800-227-2345
www.cancer.org
- Colorectal Cancer Alliance
1025 Vermont Avenue Northwest
Suite 1066
Washington DC 20005
Phone: 877-422-2030
www.ccalliance.org
- Desmoid Tumor Research Foundation
P.O. Box 273
Suffern NY 10901
Email: marlene@dtrf.org
www.dtrf.org
- Fight Colorectal Cancer
Phone: 877-427-2111
Email: info@fightcolorectalcancer.org
www.fightcolorectalcancer.org
- International Society for Gastrointestinal Hereditary Tumours (InSiGHT)
- United Ostomy Associations of America, Inc.
Phone: 800-826-0826
www.ostomy.org
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A. APC関連ポリポーシス:遺伝子およびデータベース
| 遺伝子記号 | 染色体座位 | タンパク質 | 座位特異性 | HGMD | Clin Var |
|---|---|---|---|---|---|
| APC | 5q22.2 | Adenomatous polyposis coli protein | 大腸がん遺伝子バリアントデータベース: Adenomatous Polyposis Coli (APC)
APC @ ZAC-GGM |
APC | APC |
データは以下の標準的参照資料をもとに作成した.遺伝子はHGNC;染色体座位はOMIM;タンパク質は UniProtを参照した.リンクが提供されたデータベース(座位特異性, HGMD,ClinVar)の詳細についてはこちらを参照のこと。
表B.APC関連ポリポーシスに関するOMIMの登録 (OMIMですべてをみる)
| 135290 | DESMOID DISEASE, HEREDITARY; DESMD |
| 175100 | FAMILIAL ADENOMATOUS POLYPOSIS 1; FAP1 |
| 611731 | APC REGULATOR OF WNT SIGNALING PATHWAY; APC |
| 619182 | GASTRIC ADENOCARCINOMA AND PROXIMAL POLYPOSIS OF THE STOMACH; GAPPS |
遺伝子の構造
APC遺伝子は,グリコーゲン合成酵素キナーゼ3b(GSK3B)との複合体を作る腫瘍抑制因子をコードする;この複合体は、細胞質のβカテニンを標的としてリン酸化し,その後,ユビキチンを介してプロテオソームによって分解する機能をもつ.βカテニンは,細胞接着および細胞内のシグナル伝達に関与する.APCタンパクは,細胞質でのβカテニンの蓄積を防いで正常なアポトーシスを維持し,おそらくβカテニンを制御することによって,細胞増殖を減少させている可能性がある[Zhang & Shay 2017].
また,APCは有糸分裂時に動原体に集積し,動原体-微小管接着に寄与し,染色体の安定性と分離に関与することが示されている[Fodde et al 2001, Kaplan et al 2001].APCタンパクのその他の役割としては;大腸腺窩への細胞遊走とEカドヘリンと関連した細胞接着に対する制御,GSK3Bと共に細胞極性に対する制御,その他,微小管の安定化と関連する機能を持つ [Etienne-Manneville & Hall 2003].Zhang & Shay [2017]は,APCの機能について優れたレビューを提示している.
病的なAPCバリアントは,異常な(通常は切断型)タンパクを生成し,GSK3Bと結合できないため,βカテニンを分解標的とできず,結果として,遊離の細胞質β-カテニンが高濃度で存在することになる.遊離β-カテニンは細胞核へ移動して転写因子であるTcf-4あるいはLef-1(T細胞因子-リンパ球エンハンサー因子)と結合し,がん遺伝子であるMYCやCCND1などの遺伝子発現を促進するようである[Chung 2000].異常APCタンパクは,正常な大腸腺窩の細胞配置を混乱させる可能性があり,大腸がんにおける染色体不安定性に関与すると考えられている [Fodde et al 2001].
疾患の原因となる機構.機能欠失
表7. 注目すべきAPC病的バリアント
| 参照配列 | DNA 塩基の変化 | 予測されるタンパク質の変化 | 備考[参照] |
|---|---|---|---|
| NM_000038.5 NP_000029.2 |
c.3927_3931delAAAGA | p.Glu1309AspfsTer4 | APC 生殖細胞系列病的バリアントで最も多く報告されている |
| c.3920T>A | p.Ile1307Leu | アシュケナージ系ユダヤ人の創始者バリアント [Boursi et al 2013] | |
| NM_000038.5 | c.221-1G>A | -- | ニューファンドランド島出身者の創始者バリアント [Spirio et al 1999, Woods et al 2010] |
| NG_008481.4 | Insertion of 337 bp of Alu I sequence cd. 15261 | -- | アマン派の創始者バリアント [Halling et al 1999] |
表に記載されたバリアントは著者らによって提供された.GeneReviewsのスタッフはバリアントの分類を独自に検証していない。
GeneReviews はHuman Genome Variation Society (varnomen.hgvs.org)の標準的な命名規則に従う.命名の説明に関してはQuick Referenceを参照のこと.
- 現在の命名規則に適合していないバリアント表記
がんと良性腫瘍
ほとんどの大腸腫瘍は家族性ではなく,大腸の腫瘍形成初期に起こると考えられている[Christie et al 2013, Aghabozorgi et al 2019] APCの体細胞病的バリアントが関連している[Lüchtenborg et al 2004, Christie et al 2013].更新履歴:
- Gene Review著者: Randall W Burt, MD, Kory W Jasperson, MS
日本語訳者: 櫻井晃洋(信州大学医学部遺伝医学・予防医学講座)
Gene Review 最終更新日: 2008.7.24. 日本語訳最終更新日: 2008.12.15. - Gene Review著者: Kory W Jasperson, MS and Randall W Burt, MD.
日本語訳者: 江田 肖(瀬戸病院 遺伝診療科),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Review 最終更新日: 2014.3.27. 日本語訳最終更新日: 2014.6.27. - Gene Reviews著者: Kory W Jasperson, MS, Swati G Patel, MD, MS, and Dennis J Ahnen, MD.
日本語訳者: 浦祐子(札幌医科大学大学院医学研究科遺伝医学),櫻井晃洋(札幌医科大学医学部遺伝医学)
Gene Reviews 最終更新日: 2017.2.2. 日本語訳最終更新日: 2018.5.6. -
Gene Reviews著者: Timothy Yen, MD, Peter P Stanich, MD, Lisen Axell, MS, CGC, and Swati G Patel, MD, MS.
日本語訳者: 箕浦祐子(札幌医科大学大学院医学研究科遺伝医学),櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2022.5.12. 日本語訳最終更新日: 2022.6.8[in present]
![]()