ハンチントン病
(Huntington Disease)
[Synonyms:ハンチントン舞踏病(Huntington Chorea)]
Gene Reviews著者: Nicholas S Caron, PhD, Galen EB Wright, PhD, and Michael R Hayden, MB, ChB, PhD, FRCP(C), FRSC
日本語訳者: 冨成麻帆(名古屋市立大学大学院看護学研究科)、佐橋健太郎(名古屋大学医学部附属病院 脳神経内科)、勝野雅央(名古屋大学大学院医学系研究科 神経内科学)
GeneReviews最終更新日: 2020. 6.11. 日本語翻訳最終更新日:2022.10.30.
要約
疾患の特徴
ハンチントン病(HD)は運動、認知、精神障害をきたす進行性疾患である。平均発症年齢は35-44歳であり、生存期間中央値は発症後15-18年である。
診断・検査
HDの診断は家族歴があること、特徴的な臨床所見、HTT遺伝子においてCAG三塩基繰り返し配列(リピート)の36以上の伸長の検出に基づく。
臨床的マネジメント
症状に対する治療:
薬物治療として、舞踏運動に対する、定型的な神経遮断薬(ハロペリドール)、非定型神経遮断薬(オランザピン)、ベンゾジアゼピン、モノアミン枯渇薬テトラベナジン、寡動や固縮に対する、抗パーキンソン薬、精神障害(抑うつ、精神症状、突発的な攻撃性)に対する、向精神薬や数種の抗てんかん薬、ミオクローヌス型運動過剰症に対する、バルプロ酸が含まれる。また介護需要、食事摂取、特殊な補装具に配慮した対症療法や、地方自治体や国の支援資格が挙げられる
続発性合併症の予防:
長期ケアを必要とする例に通常生じうる合併症と、薬物治療関連の副作用に注意する。
経過観察:
舞踏運動、固縮、歩行障害、抑うつ、行動変化、認知機能低下の出現および重症度を定期的に評価する。HD行動観察尺度(Behavior Observation Scale Huntington:BOSH)やHD統一評価スケール(Unified Huntington's Disease Rating Scale:UHDRS)を用いた定期的な機能評価を行う。
回避すべき薬剤・環境:
L-ドパ含有薬(舞踏運動を悪化させる恐れがある)、アルコール摂取、喫煙がある。
その他:親がHDに罹患している小児や思春期例は、地域のHD支援グループへの紹介を通じ、教材や心理的サポートの支援を得てもよい。
遺伝カウンセリング
HDは常染色体優性遺伝性疾患である。病的バリアントを有する例の子は50%の確率で疾患発症アレルを受け継ぐ。発症リスクのある無症状成人に対する発症前診断は可能であるが、現在のところ治療法がないため、慎重な検討(検査前後の遺伝カウンセリングを含む)が必要である。しかし、無症状で発症リスクのある例の中には、臨床試験の参加資格がある場合がある。18歳未満の、無症状で発症リスクある例には、発症前診断は適切でないと考えられている。また分子遺伝学的検査による出生前診断や着床前遺伝学的検査も可能である。
診断
疑わしい所見
以下のいずれかを満たす例では、ハンチントン病(HD)を疑うべきである。
- 舞踏運動を特徴とする進行性運動障害。随意運動も障害されることがある。
- 認知機能の低下、人格変化、抑うつを含む精神障害
- 常染色体優性遺伝に合致する家族歴
注:HDでは運動、認知、精神障害の発症や順序は様々である(「臨床経過」の項参照)。
診断の確立
HDの診断は、HDの臨床症状や徴候を有する発端者において、分子遺伝学的検査によるHTT遺伝子のヘテロ接合性のCAG三塩基リピートの異常伸長の同定により確定される(表1参照)。
注:臨床配列に基づくマルチジーンパネル、エクソームシーケンス、ゲノムシーケンスでは、HTT遺伝子における病原性CAGリピート伸長を現在のところ検出できない。
CAGリピートサイズ
- 正常型アレル 26CAGリピート以下。
- 中間型アレル 27-35CAGリピート。この範囲のアレルを有する場合、HD発症リスクはないが、CAG領域は不安定であるため、HD発症型域のアレルを有する子が生まれる可能性がある[Semaka et al 2006]。生殖細胞CAGリピート伸長の発症リスク推定法は確立されている[Semaka et al 2013a, Semaka & Hayden 2014]。
- HD発症型アレル 36CAGリピート以上。HD発症型アレルを1つ有する例は生涯でHDを発症するリスクがあると考えられている。HD発症型アレルは、さらに以下のように分類される:
- 低浸透性HD発症型アレル 36-39CAGリピート。この範囲のアレル保持者にはHD発症リスクがあるが、発症しない可能性もある。この範囲のCAGリピートを有する高齢の無症状者がしばしば見られる[Kay et al 2016]。
- 完全浸透性HD発症型アレル 40CAGリピート以上。このサイズのアレルは、一般的な寿命を想定した場合に、より高い確率でHD発症と関連している。
分子遺伝学的検査は、HTT遺伝子のCAGリピート数同定のための標的解析である。
表1. ハンチントン病用の分子遺伝学的検査
| 遺伝子1 | 検査法2,3 | 本法で検出される病的バリアントの発症者比率 |
|---|---|---|
| HTT | CAG三塩基リピート伸長の標的解析 | 100% |
- Table A:遺伝子と染色体座とタンパク質名データベースを参照。
- HTT遺伝子のCAGリピート数を同定する具体的な方法については表6を参照。
- 現在の臨床配列に基づくマルチジーンパネル、エクソームシーケンス、ゲノムシーケンスでは、本遺伝子の病原性リピート伸長を検出することはできない。
注:HDを予測する遺伝学的検査に関する包括的な推奨事項については、遺伝カウンセリングの項およびMacLeod et al[2013]を参照。
臨床的特徴
臨床経過
ハンチントン病(HD)の前駆期には、運動能力、認知機能、人格にわずかな変化が見られる場合がある(図1参照)[Tabrizi et al 2013, Ross et al 2014, Liu et al 2015]。これらのわずかな変化は、臨床的に明らかなHD発症の15-20年も前に起こる可能性がある。
Reilmannら[2014]は、発症前、前駆期、発症期HDの診断基準のガイドラインを提唱した;表2参照。この表は、個々を異なる診断カテゴリーに分類するために使用でき、経時的な臨床マネジメントとして有意義である。例として、発症前および前駆期HDの認識が、予防治療(対症療法ではなく)を可能とすることがある。系統分類上、遺伝学的に確定診断されたHDとの明確な相違に留意すること。
表2.ハンチントン病の診断カテゴリー
| HDの分類 | HD徴候/症状 | |
|---|---|---|
| 遺伝学的に確定 | 遺伝学的に確定していない | |
| 発症前HD:遺伝学的に確定した発症前HD | 臨床上、発症リスクのあるHD:遺伝学的に確定していないが、臨床的に発症リスクのあるHD |
|
| 前駆期HD:遺伝学的に確定した前駆期HD | 臨床上の前駆期HD:遺伝学的に確定していない前駆期HD |
|
| 発症期HD:遺伝学的に確定した発症期HD | 臨床上、発症期HD:臨床的に明らかな遺伝学的に確定していない発症期HD1 |
|
Reilmann et al[2014]より改変;許可を得て使用
DCL=diagnostic confidence level (UHDRSスケールによる診断信頼水準]);HD=Huntington disease(ハンチントン病);TFC=total functional capacity(総機能能力)
- Motor DCL=4および認知機能変化が必要
HDの平均発症年齢は約45歳である[Bates et al 2015]。約3分の2の患者は初期に神経症状を呈するが、精神症状を呈する患者もある。診断後早期には、わずかな眼球運動や協調運動の変化、軽微な不随意運動、思考困難や、しばしば抑うつ気分や興奮性などの症状が現れる(表3参照)。患者は通常、日常活動の大部分を遂行可能であり、仕事を継続可能である[Ross et al 2014, Bates et al 2015]。
HD患者の約25%は、発症が50歳以降に遅れることがあり、70歳以降に発症する場合もある。このような患者では舞踏運動、歩行障害、嚥下障害がみられるが、典型症例と比しより長期かつ良好な経過をたどる。
次の病期では舞踏運動はより顕著となり、随意運動が次第に困難となり、構音障害と嚥下障害が悪化する。大部分の患者が仕事を辞めざるを得なくなり、次第に他者の介助が必要となるが、それでもなお、かなりの部分で自立維持可能である。機能低下は大部分で著しく、攻撃的な行動や社会的脱抑制の、断続的な突発的な出現が時にみられる
HD末期では、運動障害が重度で全介助状態、無言、失禁状態となる場合が多い。発症後生存期間中央値は15-18年である(範囲:5-25年超)。死亡時平均年齢は54-55歳である[Bates et al 2015]。
表3.HDにおける臨床徴候と症状の発症
| HDの臨床徴候 | |
|---|---|
| 初期 |
|
| 中期 |
|
| 末期 |
|
運動障害 HD患者では、不随意運動と随意運動の両方に障害がみられる。非反復性、非周期的な四肢、顔面、体幹の突発的な運動からなる不随意運動障害である舞踏運動がHDの主要徴候である。舞踏運動は患者の90%以上に認められ、典型的には発症後10年間で悪化していく。舞踏運動は覚醒時に持続してみられ、意図的に抑制することができず、ストレスで増悪する。
疾患経過進行とともに、動作緩慢、固縮、ジストニアといった他の不随意運動が認められる。随意運動障害は初期徴候である。患者と家族は、日常動作のぎごちなさと表現する。動作速度、巧緻運動の制御、歩行が障害される。眼球運動障害も初期に起こり、徐々に悪化する。衝動性、緩徐衝動性および水平衝動性の眼球運動の開始困難、注視固定障害が、症候性患者の75%に認められうる[Blekher et al 2006, Golding et al 2006]。構音障害は、初期によく認められる。嚥下障害は、末期に起こる。腱反射亢進は初期に90%の患者にみられるが、クローヌスや伸展性足底反射は後期でみられ、出現頻度は少ない。
認知障害 認知能力の全般的進行性低下が全てのHD患者で起こる。認知能力の変化には、物忘れ、思考遅延、視空間能低下および習得知識の処理能力低下がある。運動症状の発症以前に、軽微ではあるが明らかな認知障害が認められたとする研究がいくつかある[Bourne et al 2006, Montoya et al 2006, Paulsen et al 2008, Tabrizi et al 2009, Rupp et al 2010]。初期の変化は思考柔軟性の欠如や、思考や活動の連続性構築などの実行障害を伴うことが多い。
情報想起の多大な障害を伴う記憶障害が初期に起こるが、言葉の手がかり、プライミング効果、十分な時間的猶予が、部分的もしくは正確な想起につながることもある。HD初期の記憶障害は、通常アルツハイマー病と比し重症度はかなり下がる。
HD患者の認知および行動症状は概ね、アルツハイマー病より前頭側頭型認知症に類似している。注意、集中力は早期に低下し[Peinemann et al 2005]、注意散漫となる。言語機能は比較的保たれるが、後期には統語的複雑レベルの低下、皮質性発話障害、錯語、喚語困難がよくみられる。
神経心理学的検査では、特に疾患後期において視空間能力障害が示される。特に自分自身の障害に対して自覚が欠如していることが多い[Ho et al 2006, Bates et al 2015]。
精神障害 HD患者には著しい人格変化、情動性精神症、統合失調性精神症が生じる[Rosenblatt 2007]。HD発症以前の抑うつ、敵意、強迫性、不安、精神症気質の測定スコアは高い傾向にある[Duff et al 2007]。進行性認知運動障害とは異なり、精神的変化は疾患の重症度に伴って進行しない傾向がある[Epping et al 2016]。間欠性突発症、無気力、攻撃性、アルコール濫用、性機能障害や性的逸脱、食欲亢進といった行動障害がよくみられる。妄想の多くは被害妄想であり一般的である。幻覚はそれほど多くない。
抑うつと自殺リスク 発症前および症状のある患者では、一般的な集団と比べて抑うつ症状の頻度は2倍以上である[Paulsen et al 2005b, Marshall et al 2007]。HDにおける抑うつの発症原因は不明であるが、HDを発症したことによる心理的影響ではなく病態そのものによる可能性がある[Slaughter et al 2001, Pouladi et al 2009]。HD患者の自殺や自殺企図は多いが、疾患の経過や発症前診断の結果により、その発生率は変化する[Larsson et al 2006, Robins Wahlin 2007, van Duijn et al 2018]。自殺リスクが高い時期は、診断を受ける直前と、のちに患者が自立不可となる時であることが分かっている[Baliko et al 2004, Paulsen et al 2005a, Eddy et al 2016]。
その他 HD患者の肥満度指数(BMI)は対照群と比べて低く[Pratley et al 2000, Stoy & McKay 2000, Djoussé et al 2002, Robbins et al 2006]、代謝変化と相関している可能性があり[Duan et al 2014]、臨床的進行のバイオマーカーとなり得る[van der Burg et al 2017]。また、HDの患者はコレステロール代謝障害を示す[Valenza & Cattaneo 2006, Wang et al 2014]。HD患者が食欲とエネルギー消費量増大を示すことも一般的である[Pratley et al 2000, Trejo et al 2004, Gaba et al 2005]。
HD患者では、睡眠と概日リズムはが乱れており[Goodman & Barker 2010, Morton 2013]、おそらく視床下部の機能障害[Petersén & Björkqvist 2006]やメラトニン分泌の変化[Kalliolia et al 2014]の結果である可能性がある。不眠や日中の傾眠もみられる場合もあるが、一般的には精神的変化、抑うつ、もしくは舞踏運動がより原因となることが多い[Videnovic et al 2009]。
神経病理学 HDの神経病理学的特徴は、尾状核と被殻および大脳皮質における神経細胞の変性である[Waldvogel et al 2015]。大脳基底核の運動制御の間接路の、エンケファリン含有中型有棘神経細胞の優先的変性が、舞踏運動の神経生物学的基盤をもたらす[Galvan et al 2012]。さらに、直接路の、サブスタンスP含有中型有棘神経細胞が失われると、無動とジストニアが発生する[Galvanetal2012]。また淡蒼球、視床下核、視床、視床下部、黒質、海馬の神経細胞消失に関するエビデンスもある[Vonsattel et al 1985, Vonsattel & DiFiglia 1998, Heinsen et al 1999, Petersén et al 2005, Guo et al 2012, Domínguez et al 2013, Singh-Bains et al 2016]。大脳基底核と皮質における神経細胞消失の領域特異的なパターンが、患者の最も顕性化した症状の基盤となり、個人間の表現型の多様性に寄与する可能性がある[Thu et al 2010, Hadzi et al 2012, Kim et al 2014, Waldvogel et al 2015, Mehrabi et al 2016]。また、末梢組織に病理所見が認められることもある[Björkqvist et al 2008, van der Burg et al 2009]。
HTT遺伝子の発現タンパク質であるhuntingtin(ハンチンチン)含有神経細胞内封入体も、HDのよく知られた神経病理所見である。しかし、huntingtinタンパク質の発現や脳内でのhuntingtin含有封入体のパターンと出現時期は、疾患の選択的変性と相関しなく、主要な病態決定因子と考えられていない[Kuemmerle et al 1999, Michalik & Van Broeckhoven 2003, Arrasate et al 2004, Slow et al 2005, Slow et al 2006]。
神経画像 画像検査はHDの臨床診断をさらに支持し、疾患の進行の検査のための有用な手法である[Biglan et al 2009, Paulsen 2009, Tabrizi et al 2011, Tabrizi et al 2012, Tabrizi et al 2013]。症状のみられる患者での有意な線条体萎縮に加えて、局所的および脳全体の灰白質および白質の変化も検出されている[Majid et al 2011, Tabrizi et al 2011, Tabrizi et al 2012, Tabrizi et al 2013]。さらに、MRI検査により、発症予測時より何年も前に進行性の灰白質および白質萎縮が確認されている[Tabrizi et al 2011, Tabrizi et al 2012, Tabrizi et al 2013]。近年、神経画像を用いてHDの臨床的進行を解明する研究が数多く行われており、実験的治療法の有効性検証の臨床試験に、以上のような客観的指標の使用に特に関心が持たれている[Tabrizi et al 2012, Tabrizi et al 2013]。
若年性HD 若年性HDは20歳以前の症状発現と定義され、HD患者の5-10%を占める[Gonzalez-Alegre & Afifi 2006, Quarrell et al 2013]。成人HD患者で観察される運動、認知、精神機能障害は若年性HD患者にも認められるが、表現型は異なる。重度の精神症状悪化、顕著な運動・小脳症状、言語発達遅滞、急速な悪化も若年性HDの特徴である[Nance & Myers 2001, Gonzalez-Alegre & Afifi 2006, Squitieri et al 2006, Yoon et al 2006]。若年発症群だけにみられるてんかん発作は、発症年齢が10歳以前のHD患者の30-50%に認められる[Gonzalez-Alegre & Afifi 2006]。
思春期発症型では、症状が成人型により類似し、通常、舞踏運動や重度の行動障害が初期症状としてみられる[Nance & Myers 2001]。
中間型アレル CAGリピート数が27-35の例では、HD発症リスクはないと考えられているが、CAGリピートの不安定性のため、病因CAGリピートのアレルを子が有するリスクはある可能性がある[Semaka et al 2006, Kay et al 2018]。限られたデータではあるが、中間型アレルを有する例が行動変化および運動・認知障害を呈することが示唆されているが、この点に関してはさらなる検証が必要である[Killoran et al 2013, Cubo et al 2016]。
遺伝子型・臨床型相関
CAGリピート数とHD発症年齢の間には、有意な逆相関関係が存在する[Langbehn et al 2004, Langbehn et al 2010]。分子遺伝学の項参照。
- 成人発症患者では、通常36-55CAGリピートのHTTアレルが認められる。
- 若年発症患者では、通常60以上のCAGリピートのHTTアレルが認められる。
- 中間型アレル(27-35CAGリピート)は通常疾患との関連はないが、CAGリピートの不安定性を受けやすい [Semaka et al 2013b]
三塩基リピート数による年齢別発症予測に関するデータについてはubc.ca (pdf).を参照。
臨床的発症年齢に加え、CAGリピート数も死亡年齢を予測すると示されたが、罹患期間の予測はされていない[Keum et al 2016]。
CAGリピート数が長いほど、運動、認知、機能評価項目の悪化度が増加する[Aziz et al 2009, Chao et al 2017]。
行動症状の進行は、リピート数とはおそらく相関しない[Ravina et al 2008]。
完全浸透性HDアレルのホモ接合体例は、おそらくヘテロ接合体例と同様の発症年齢であるが、疾患進行は加速している可能性がある[Squitieri et al 2011, Lee et al 2012]。
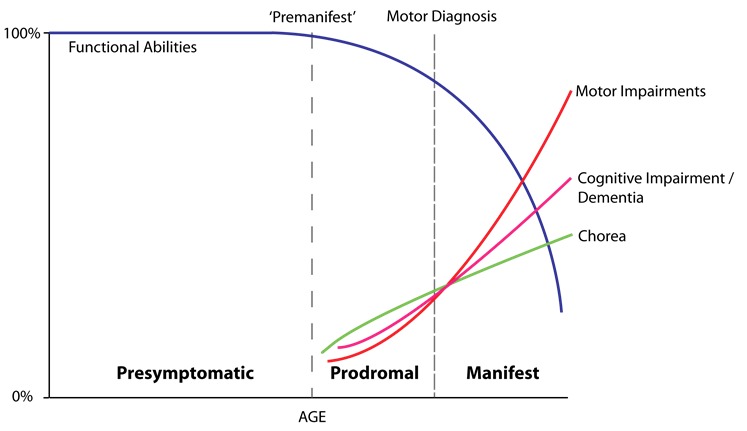
図1.ハンチントン病(HD)の自然歴。発症前の例にはHD徴候や症状はない。前駆期には通常、運動症状の有無に基づくHD診断に至るまでの間に、微細な徴候や症状が認められることがある。HD顕性期では、舞踏運動が最もよくみられる特徴の1つであり、その後、運動・認知障害の緩徐進行が続いておこる。
Ross et al[2014], Nature Reviews Neurologyを改変; Macmillan Publishers Ltdの許可を得て転載
CAG長と発症年齢の多彩性の間にも有意な負の相関が存在し、遅発性の発症年齢の多彩性はCAG長が小さいほど大きく、CAG以外の疾患修飾因子はCAG長が大きい場合と比べて、小さい場合により影響を与えることが示唆されている[Langbehn et al 2004, Gusella & Macdonald 2009]。平均して、CAGリピート長が発症年齢の多彩性に対し最大70%関与し、残り10-20%には遺伝的要因の関与が見積もられている[Li et al 2006, Gusella & Macdonald 2009]。他遺伝子座の多くの遺伝子の、少ないながらもこの遺伝的多彩性への関与が示された[GeM-HD Consortium 2015]。
近年、HTT遺伝子座およびゲノム全体に存在する、これらの追加のゲノム修飾因子同定の大きな進展があった。
- シス作用性因子 最も一般的なCAGリピート(アレルの95%以上)は、最後から2番目のリピート[(CAG)n-CAA-CAG]部分でCAA中断を有する。中断のないCAGリピート[(CAG)n]は、HD発症年齢の低下およびリピート不安定性の増大と関連する[Ciosi et al 2019, GeM-HD Consortium 2019, Wright et al 2019](GeM-HD 2019 full text)。これらの知見は、HDの推定発症年齢の予測に対し、ポリグルタミン長よりも中断のないCAG長の方が優れていることを示唆している。
- トランス作用性因子 ゲノムワイド関連研究により、HDの他の修飾遺伝子候補が同定された[GeM-HD Consortium 2015, Moss et al 2017]。これらの解析により、DNA修復、ミトコンドリア分裂、酸化還元酵素活性関連の生物学的経路が表現型に重要な役割を果たすことが示され、今後の研究のための修飾遺伝子候補が同定された(例としてFAN1, MLH1, MTMR10, MSH3, およびRRM2B)。
浸透率
36-39CAGリピートアレルは、HD発症アレルと考えられているが、不完全浸透を示す。この範囲のCAGリピートを有する高齢の無症状例がしばしばみられる[Kay et al 2016]。
疾患リスクは、アレルの95%以上に観察される一般的な[(CAG)n-CAA-CAG]中断リピートと、アレルの約1%に観察される稀な[(CAG)n]非中断リピートと間で異なる[Ciosi et al 2019, GeM-HD Consortium 2019, Wright et al 2019](GeM-HD 2019 full text):
- 36-39リピート 発症者の約3分の1が純粋な[(CAG)n]リピートを有する。
- 36および37リピート 発症者の大多数は純粋な[(CAG)n]リピートを有する。
CAAリピートの消失はゲノム刷込みの消失(LOI)バリアントと呼ばれ[Wright et al 2019]、36-39CAGリピートの例での発症率に影響し得るであろう。
40以上のCAGリピートを含むアレルは完全に浸透する。40以上のCAGリピートのアレルを有し、無症状である高齢者例の報告はない。
表現促進現象
HDでは、世代を経るごとに重症化し、発症年齢が若年化する表現促進現象が起こると知られている。表現促進現象は、変異アレルを父親から受け継いだ場合にはるかに多く起こる。表現促進現象は、精子形成時の不安定なCAGリピートに起因する[Semaka et al 2013a]。大規模な伸長(すなわち、CAGリピートが7を超えるアレルサイズの増加)は、ほとんどが父親由来の遺伝により起こる。大多数の若年発症の患児は、父親より伸長アレルを受け継ぐが、時に母親から受け継ぐこともある[Nahhas et al 2005]。
命名法
分子遺伝学が登場する前には、聖ヴィート(St. Vitus's dance)舞踏病、シデナム(Sydenham's)舞踏病など、舞踏病には様々な病名があった。
若年発症HDあるいは小児発症HDは、以前からウェストファル(Westphal)型HDと呼ばれていた。
まだ症状が認められていない人はHD症状前期にある。舞踏運動および、または他の症状が確認された例は、明らかなHDである。
有病率
HD有病率は世界の地域によって異なる。ヨーロッパに祖先ルーツを持つ集団は10万人あたり9.71人の平均有病率を示す[Rawlins et al 2016]が、最高推定10万人あたり17人と報告されている[Fisher & Hayden 2014, Baig et al 2016]。一方、日本、中国、韓国、フィンランド、および南アフリカのアフリカ先住民族では、HDははるかに少なく、推定有病率は10万人あたり0.1人から2人とされる[Pringsheim et al 2012, Sipilä et al 2015, Xu & Wu 2015]。
ベネズエラのマラカイボ湖地域の住人は、世界で最も高いHD有病率を有すると考えられている[Wexler et al 2004]。
HDの患者分布が不均一であることは、これらの民族の一般集団における特異的な素因アレルとハプロタイプの分布によって少なくとも部分的に説明される[Warby et al 2009, Warby et al 2011, Kay et al 2018]。例として、15の種々の国際集団を対象とした最近の研究では、集団の平均CAGリピート長、および中間型アレル頻度がHD有病率と相関し、主要な祖先集団間の有病率の違いに寄与していることが示された[Kay et al 2018]。
不完全浸透HTTアレル(診断の確立、CAGリピートサイズの項参照)は、近年、一般集団に高率で存在し、400人に1人もの例で有していることが示された。ただし、浸透率は既報告より低いことが判明した(すなわち、36-38CAGアレルで0.2-2%)[Kay et al 2016]。
遺伝的関連(アレル)疾患
HTTの両アレル型病原性ミスセンス変異は、常染色体劣性遺伝の神経発達異常疾患であるロペス・マシエル・ロダン(Lopes-Maciel-Rodan)症候群(LOMARS)(OMIM 617435)を引き起こすと報告されている。LOMARSの患者は、小児期にレット(Rett)症候群に類似した表現型と知的障害を呈す。
鑑別診断
ハンチントン病(HD)は舞踏病、認知症、精神障害の鑑別診断に該当する。いくつかのHD類似疾患の鑑別診断は本誌で要約され、また別で総説されている[Schneider et al 2007, Martino et al 2013]。またアルツハイマー病とHDの併発も報告されている[Davis et al 2014]。
非遺伝性疾患も舞踏運動を伴うが、HDが疑われる患者では関連する所見と疾患経過から、ほとんどの疾患は容易に除外可能である。舞踏運動の原因として遅発性ジスキネジア、レボドパ誘発性ジスキネジア、甲状腺中毒症、脳血管疾患、中枢神経ループス、赤血球増加症、A群β溶血性レンサ球菌が含まれる。
遺伝性病態 表4を参照。
表4.ハンチントン病の鑑別診断で考慮すべき遺伝性疾患
| 疾患 | 遺伝子 | 遺伝形式 | 臨床的特徴 | |
|---|---|---|---|---|
| HDと同様の症状 | HDと鑑別すべき症状 | |||
| 前頭側頭型認知症 および/または筋萎縮性側索硬化症 | C9orf72 | AD |
|
|
| ハンチントン病類縁疾患1型 (HDL1)(OMIM 603218)1 | PRNP | AD | HDと同様の様々な臨床的特徴 |
|
| ハンチントン病類縁疾患2型 (HDL2) | JPH3 | AD | 臨床的にHDと鑑別困難 | アフリカ系において最高の有病率、またはアフリカ系限定と推定 |
| 有棘赤血球舞踏病(ChAc) | VPS13A | AR |
|
|
| マクロード神経有棘赤血球症候群 (MLS) | XK | XL |
|
|
| 脊髄小脳失調症(SCA17) | TBP | AD |
|
小脳性運動失調が主要運動障害 |
| 歯状核赤核淡蒼球ルイ体萎縮症(DRPLA) | ATN1 | AD |
|
運動失調とミオクローヌスが主要運動障害 |
| 良性遺伝性舞踏病(OMIM 118700) | NKX2-1 | AD | 舞踏運動 |
|
| 遺伝性小脳失調症(概説「運動失調症」参照) | 多数 | AD AR XL |
運動障害 | 著明な小脳・錐体路徴候を伴う遺伝性小脳失調 |
| 家族性クロイツフェルト・ヤコブ病(fCJD)(概説「遺伝性プリオン病」参照) | PRNP | AD |
|
|
| 早期発症型家族性アルツハイマー病 | APP PSEN1 PSEN2 |
AD | 認知症 | 運動障害はない |
| 17番染色体に連鎖する家族性前頭側頭型認知症パーキンソニズム | MAPT | AD |
|
舞踏運動はみられない |
AD = autosomal dominant(常染色体優性遺伝);AR = autosomal recessive(常染色体劣性遺伝);CNS = central nervous system(中枢神経系);MOI = mode of inheritance(遺伝様式);XL = X-linked(X連鎖)
1.HDL1は第20番染色体短腕上のプリオンタンパク(PrP)遺伝子であるPRNP遺伝子の特異的病的バリアント(8つの過剰なオクタペプチドリピート)により発症する[Laplanche et al 1999, Moore et al 2001]。同遺伝子座の類似病的バリアントは、家族性クロイツフェルト・ヤコブ病といったプリオン病という他疾患も引き起こす。
小児HDの診断は、HDの家族歴のある家系では確実である。孤発例(HD家族歴のない家系で唯一の罹患者)では、毛細血管拡張性運動失調症、パントテン酸キナーゼ関連神経変性症(従来ハラーホルデン・スパッツ(Hallervorden-Spatz)症候群として知られていたもの)、レッシュ・ナイハン(Lesch-Nyhan)症候群、ウィルソン(Wilson)病、進行性ミオクローヌスてんかん[Gambardella et al 2001]、その他の代謝疾患を除外する必要がある。
臨床的マネジメント
初期診断後の評価
ハンチントン病(HD)と診断された患者の疾患の重症度と医療ニーズの確定に、本章で要約した評価(診断に至った評価の一部として行われていない場合)が推奨される:
- 身体学的検査
- 神経学的評価
- HD関連の運動、認知、精神症状の全般評価。前述の臨床尺度システムのうち、ハンチントン病統一スケール(Unified HD rating scale:UHDRS)が、HDの臨床的特徴と進行に関する、信頼性の高い安定した評価を可能とする。
- 臨床遺伝学者や遺伝カウンセラーへのコンサルテーション
対症療法
薬物治療は対症療法に限られる[Mestre et al 2009, Killoran & Biglan 2014]。
- 定型神経遮断薬(ハロペリドール)および非定型神経遮断薬(オランザピン)、ベンゾジアゼピン、モノアミン阻害薬であるテトラベナジンによって舞踏運動を部分的に抑制することができる[de Tommaso et al 2005, Bonelli & Wenning 2006, Huntington Study Group 2006]。テトラベナジンは、舞踏運動に対する治療薬として有効であるが、その使用は錐体外路症状などの重篤な副作用につながる。テトラベナジンの重水素化アナログであるデューテトラベナジンは、分子の特定部位での重水素原子置換によって改良され、半減期と全身分布を向上させている[Stamler et al 2013]。これらの特性により、より少ない副作用でより頻度の少ない投与が可能となる[Frank et al 2016, Reilmann 2016]。
- 抗パーキンソン病薬は、運動減少や固縮を改善することがあるが、舞踏運動を増加させることがある。
- 抑うつ症状、精神症状、易怒性といった精神障害には一般に向精神薬やある種の抗てんかん薬が奏効する
- バルプロ酸によってHDのミオクローヌス型運動過剰症が改善された[Saft et al 2006]。
介護、栄養、特殊装具、国や地方自治体の給付受給資格に配慮した生活支援により、HD患者とその家族は恩恵を受けることができる。数々の社会的問題がHD患者とその家族に付きまとうが、実用的な支援、精神的サポート、カウンセリングにより軽減される[Williams et al 2009]。
続発的合併症の予防
主なHDの続発的合併症には下記が含まれる:
- 長期的な対症療法を要する患者で典型的に起こる合併症
- 様々な薬物療法に関連した副作用。薬物の副作用は,関連薬物、投与量、個人因子を含む様々な要因に依存する。通常、HDで用いられる薬剤では、副作用は抑うつ、鎮静、悪心、落ち着きのなさ、頭痛、好中球減少症、遅発性ジスキネジアを含むことがある。一部の患者では、ある種の治療の副作用が症状よりひどい場合もある。そのような患者には、治療の中止、投与量の減量、定期的な休薬が良いことがある。舞踏運動治療に用いられている現薬剤は、特に重篤な副作用を起こしやすい。軽度から中等度の舞踏運動の患者に対しては、動作訓練や言語治療など非薬物療法の支援がより良い場合がある。
- 抑うつ症状が見られる場合は、標準治療が適している[Paulsen et al 2005b, Phillips et al 2008]。
経過観察
定期的な評価により、舞踏運動、固縮、歩行障害、抑うつ、行動変化、認知機能低下の出現や重症度の検討を行う[Anderson & Marshall 2005, Skirton 2005]。
ハンチントン病行動観察スケール(Behavior Observation Scale Huntington:BOSH)は,介護施設環境下のHD患者の機能的能力を迅速かつ長期的に評価するために開発されたスケールである[Timman et al 2005]。縦断研究にはハンチントン病統一評価スケール(Unified HD rating scale:UHDRS)が用いられる[Huntington Study Group 1996,、Siesling et al 1998, Youssov et al 2013]。全機能的能力(total functional capacity:TFC)スケールはHDの進行、患者の機能レベル、介護者の追加援助の必要度を表すのに使用される。
回避すべき薬剤/環境
L-ドパ含有化合物は、舞踏運動を増悪させる場合がある。
禁酒と禁煙が勧められる。
リスクのある血縁者の評価
遺伝カウンセリング目的のリスクのある血縁者の検査に関連する問題は、「遺伝カウンセリング」の項参照。
研究開発中の治療法
多様な候補治療法が、HDの動物モデルやヒトを対象とした臨床試験の両方にて検証中である[Wild & Tabrizi 2014]。この多様性は、HDで障害されることが知られる様々な細胞経路を反映している[Bonelli et al 2004, Rego & de Almeida 2005, Borrell-Pagès et al 2006, Graham et al 2006, Bonelli & Hofmann 2007]。
- 検証中の薬剤には、アポトーシス、興奮毒性、huntingtinタンパク質凝集・分解・リン酸化、炎症、酸化障害、ホスホジエステラーゼ活性、ヒストン脱アセチル化酵素、トランスグルタミナーゼ活性の阻害剤や、ミトコンドリア機能、分子シャペロン活性、転写、神経栄養の調節化合物がある。
- 非臨床HD動物モデルで改善が示され臨床試験に進められている治療法は、ミノサイクリン、酪酸ナトリウム、必須脂肪酸、ラセミド、クレアチン、シスタミン、リルゾール、メマンチン、レスベラトロール、大麻抽出物(THCおよびCBD)、コエンザイムQ10、ホスホジエステラーゼ10a阻害剤がある[Bender et al 2005, Puri et al 2005, Bonelli & Wenning 2006, Ondo et al 2007, Okamoto et al 2009, Huntington Study Group DOMINO Investigators 2010, Hersch et al 2017, McGarry et al 2017]。注目すべきは、これら治療化合物の多くが、臨床評価項目の到達とHDの臨床的進行を修正する効力の実証に失敗している。プリドピジン、ラキニモド、セマフォリン4D中和抗体など実験的治療化合物は、未だ臨床開発途上である。
- HD原因を標的とする遺伝子サイレンシングは、非臨床動物試験において安全かつ有効であることが示されており、現在で臨床試験中または開始途中にある。これらには、RNA干渉(RNAi)またはアンチセンス核酸(ASO)を用いたアプローチが含まれる[Boudreau et al 2009, Pfister & Zamore 2009, Hu et al 2010, McBride et al 2011, Kordasiewicz et al 2012, Aronin & DiFiglia 2014, Dufour et al 2014, Stanek et al 2014, Pfister et al 2018]。これらのアプローチは、アレル非選択的な方法で全huntingtin発現を抑制することを目的とするか[Boudreau et al 2009, McBride et al 2011, Kordasiewicz et al 2012]、または変異HTTアレルのみに対して選択的とする[Gagnon et al 2010, Carroll et al 2011, Evers et al 2011, Skotte et al 2014, Southwell et al 2014, Datson et al 2017]、いずれかである。アレル選択性は、異常伸長CAGリピートを標的[Gagnon et al 2010, Evers et al 2011, Datson et al 2017]、または異常伸長CAGとリンクした連鎖不平衡における遺伝子多型を標的とし[Carroll et al 2011, Skotte et al 2014, Southwell et al 2014, Kay et al 2015, Southwell et al 2018]、達成可能である。HTT標的ASOを評価する最初のヒト第I/IIa相臨床試験の結果は、IONIS-HTTRx(RG6042)が試験した全ての検証用量で忍容性が高く、髄液中の用量依存的な変異HTTの抑制をもたらすことを示した[Tabrizi et al 2019]。
- HDの細胞移植試験は少数例に対し様々な結果が示された[Furtado et al 2005, Bachoud-Lévi et al 2006, Farrington et al 2006, Dunnett & Rosser 2007, Barker et al 2013]が、追加の大規模研究が進行中である[Lopez et al 2014]。さらに、静脈注射された間葉系幹細胞の安全性と有効性について現在ヒト初の臨床試験(NCT02728115)にて、HD患者を対象に検証されている。懸念事項として、近年の研究で、変異huntingtinが同種移植された神経組織に拡散伝播する能力があることが示唆されている[Cicchetti et al 2014]。
HDに対し数多くの臨床試験が計画または進行中であり、huntingtonstudygroup.orgに掲載されている。数多くの薬物試験が完了し、あるいは現在進行中である。
米国ではClinicalTrials.gov、ヨーロッパではEU Clinical Trials Registerでの検索により、様々な疾患や病状に関する臨床試験の情報が入手可能である。
その他
TRACK-HD、PREDICT-HD、ENROLL-HDなどのバイオマーカー研究は、画像、臨床スケール、生理的測定値を用いて疾患進行の初期変化を特定するために実施されている[Scahill et al 2012, Ross et al 2014]。HD有リスク者の縦断研究も行われている[Huntington Study Group PHAROS Investigators 2006, Paulsen et al 2006, Tabrizi et al 2012]。
HD患者コホートにおいて、疾患発症と臨床的進行の分子バイオマーカー候補が数多く評価されているが、有用性が確認されているものはごくわずかである。髄液中の変異huntingtinレベルは、HDの病期と相関することが示されており[Southwell et al 2015, Wild et al 2015]、脳内の変異huntingtinレベルを反映している可能性がある[Southwell et al 2015]。したがって髄液中の変異huntingtinレベルは、臨床的進行の確かなバイオマーカーとなり、HTT標的臨床試験のための脳内HTT抑制の代替測定法となる可能性がある。さらに血液および髄液中のニューロフィラメント軽鎖のレベルは、HD患者における疾患発症や臨床的進行、および局所脳萎縮の予後バイオマーカー候補となることが示されている[Byrne et al 2017, Johnson et al 2018]。
HD患者の親を持つ小児や青年は時に非常に恵まれない環境にあり、特殊な問題を抱えることがある。教材や必要とされる心理的サポートを受けるためのHD地域支援グループへ紹介が有益である(「リソース」の項参照)。
アルツハイマー病治療に用いられるドネペジルは、HDの運動機能や認知機能を改善しない[Cubo et al 2006]。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ハンチントン病(HD)は、常染色体優性遺伝性疾患である。
家族構成員のリスク
発端者の両親
- 大多数HD診断例は、罹患した親をもつ。
- 数名のHD診断例のある家族歴が、以下一事由により、ないと見なされる場合がある:
- 親族の疾患を認識していない場合
- 症状発症前の親の早期死
- 無症状の親が中間型アレル(27-35CAGリピート)もしくは低浸透性HTTアレル(36-39CAGリピート)を有する場合(「発端者の子孫」の項参照)
- 罹患者である親が遅発型である場合
- 分子遺伝学的検査は、単独例(すなわち、家系内での単一発症例)を表すとみなされる発端者の両親に対し推奨される。
発端者の同胞
発端者の同胞が有するリスクは、発端者の両親の遺伝状況に依存する:
- アレルのCAGリピートサイズ CAGリピートサイズが大きいとより伸長しやすい。 精子におけるCAGリピートサイズ別の不安定性の推定は、中間型アレルの予測検査の結果を受け取ったケースに対し、遺伝カウンセラーからの、より正確なリスク評価の提供を可能とすると報告されている[Hendricks et al 2009, Semaka et al 2013a, Semaka & Hayden 2014]。全ての中間型CAGリピートサイズが伸長する可能性を有することが示されたが、伸長発生率はCAGリピートサイズが大きくなるにつれて劇的に増加し、35CAGリピートアレルの約21%が疾患関連範囲に伸長した。中間型アレル予測検査結果に対するエビデンスに基づく遺伝カウンセリング意義についてはSemaka & Hayden[2014]により報告されている。
- 親の性別と年齢 父方遺伝の中間型アレルは、母方遺伝の中間アレルよりもCAGリピートの伸長がおこりやすく、母方のリピート伸長は極めて稀である[Semaka et al 2015]。伸長した中間長アレルは、高齢時の父親から受け継がれやすい。
- CAGリピート伸長を有するDNAシス配列 CAAやCCG三塩基で中断されたCAGリピートは、より安定となる。
発端者の子
- HTTにおけるCAGリピート伸長のヘテロ接合の結果として、HD患者の子は受精時にHD原因アレルを50%の確率で受け継ぐ。受け継ぐ子は下記の通りである。
- 低浸透性アレル(36-39CAGリピート)はHDのリスクがあるが、発症しないこともある(「浸透率」の項参照)。
- 完全浸透性アレル(40以上のCAGリピート)は、正常寿命と仮定した場合、より高い確率でHD発症のリスクがある。
- HTTのCAGリピート伸長がホモ接合である患者の子は、HD原因アレルを受け継ぐことになる。
他の家族構成員
他の血縁者のリスクは発端者の両親からの遺伝状況に依存する。親が罹患している、またはHTTのCAGリピート伸長を有する場合、その血縁者にリスクがある。
遺伝カウンセリングに関連した問題
予測検査(すなわち、無症状でリスクのある例の検査) HDリスクのある無症状成人に対し、検査可能である。明確な疾患症状がない場合に病的バリアント検査は、予測検査である。このような検査は、無症状の例の発症年齢、重症度、症状の種類、進行速度の正確な予測には有用でない。しかし、Langbehnら[2004]、[2010]によって報告された、特定のCAGリピートサイズを有する例が、ある種の年齢で発症する可能性に関するデータは有用であろう。現在の診断検査は、標準的な配列における最後から2番目のCAAまでのCAGリピート長までしか測定することができない。中断欠損バリアント(CAAからCAGへの置換)が存在する場合、現行の診断方法ではCAGリピート長を2リピート分、過小評価するであろう。このことは、大多数のHD患者では影響しない[Wright et al 2019]。ubc.ca(pdf)の付録表参照。HDリスクがある例を検査する場合、血縁者の疾患がHDであることを確認するために、罹患した血縁者でHTTのCAGリピート伸長を検査することは有用である。
- リスクのある無症状の成人血縁者は、出産、経済的問題、キャリアプランに関する個人的な意志決定をする上で検査を希望することが可能である。リスクある無症状例は、臨床試験参加の対象資格がある場合もある。その他、単純に「知る必要がある」といった別の動機を持つ場合もあろう。リスクある無症状の成人血縁者の検査では、通常、検査前に面接があり、検査を希望する動機、HDに関する個々の知識量、検査の陽性および陰性結果の影響について話し合いが行われ、神経学的状態も評価される。検査を希望する者は、健康、生活、障害保険適用、就労や就学上の差別、社会や家族との関係変化に関して直面しうる問題について、カウンセリングを受けるべきである。興味深いことに、ある研究では、遺伝子検査は差別リスクを増加させないことが分かった。遺伝的差別を感じるのは、HD遺伝子検査の具体的な結果よりも、遺伝状況の有無に関わらずHDの家族歴に起因する可能性がより高いことが分かっている[Bombard et al 2009]。考慮すべきその他問題として、他の血縁者のリスク状態への影響がある[Bombard et al 2012]。うつ病と希死念慮は、HDの予測検査プログラムの一部として取り組むべき問題である[Robins Wahlin et al 2000, Robins Wahlin 2007]。インフォームドコンセントを取得し、記録は機密扱いにしなくてはならない。変異アレル保持者は、長期的フォローアップと定期的評価ための整備を要する。
- カナダ発症前検査プログラム(Canadian Predictive Testing Program)参加者に対する短期期間フォローアップで、HD発症予測検査は、一部では負の経験があったにも関わらず、リスクのある例の心理的健康を維持、もしくはむしろ改善させることが示された。リスク低下が確定したグループの約10%は、この新たな状況への適応に深刻な困難を抱えていた。このような例にとっての大きな問題は、計画の立たない将来に直面しているという自意識である。全体として、リスクのある成人無症状例の検査に対する需要は、直接的な分子遺伝学的検査が利用可能になる以前に行われた研究で期待されていたより低かった。一般的な医療サービスや遺伝子検査の利用と同じく、女性は男性よりもHD発症の予測検査を受ける傾向にある[Taylor 2005, Baig et al 2016]。
- HD原因アレルを受け継いだ無症状の例のパートナーの心理的苦痛に関する研究において、Decruyenaereら[2005]は、HD原因アレルを有することが判明した例と、少なくとも同じ程度の苦痛をパートナーが有し、しかも彼らの苦悩は「剥奪状態」とされ、社会に認知されない傾向にあることを見出している。
未成年者の発症予測検査(すなわち、18歳未満でリスクのある無症状の例の検査)
- 早期治療が疾患の罹患率や死亡率に有益な効果をおそらく及ぼすことがなく、成人発症状況のリスクを有する無症状未成年者にとって、予測遺伝子検査は、主に説得力のある利益のない未成年者の自主性を否定しているため、不適切と考えられている。さらに、そのような情報がもたらす、家族の関係性、将来の差別や汚名リスク、不安への不健康な悪影響の可能性が懸念される。
- 詳細については、National Society of Genetic Counselorsの、成人発症疾患に対する未成年者の遺伝学的検査に関する立場表明、American Academy of Pediatrics and American College of Medical Genetics and Genomicsの提言:小児の遺伝学的検査およびスクリーニングにおける倫理的問題と方針を参照すること。
ハンチントン病の診断がついた家系の、症状のある例に検査を考慮するのは年齢にかかわらず適切である。
明らかなde novo病的バリアントを有する家族における配慮 両親のいずれもがHDの原因となるHTTアレル(>35CAGリピート)または中間型アレル(27-35リピート)を有さない場合、生物学的父親もしくは母親が異なる場合(例として生殖補助医療による)および未公開の養子縁組を含む非医学的な原因の調査が行われることがある。
家族計画
- 遺伝的リスクの判定や出生前/着床前遺伝学的検査の話し合いの最適時期は、妊娠前である。
- 同様に、無症状でリスクある血縁者の遺伝状況を確定する検査の意思決定も、妊娠前に行うことが最善である。
出生前検査・着床前遺伝学的検査
リスク50%のある胎児 HDの原因となるHTTアレルが、発症した親か、リスクがある親の血縁者で確定された場合、リスクの高い妊娠に対する出生前検査が可能である。
着床前遺伝学的検査(PGT)は、HDの原因となるHTTアレルが罹患血縁者にて同定された家族には、着床前遺伝学的検査(PGT)が選択肢となりうる。既存のPGT除外プロトコールは、HDの原因となるアレルの発症前検査を望まない、発症リスクのある家族におけるカップルの受精卵検査を可能としている[Sermon et al 2002, Stern et al 2002, Moutou et al 2004, Jasper et al 2006]。HDのPGTに関するカウンセリングや倫理的問題には、Asscher & Koops[2010]により議論されている。
妊娠中絶あるいは早期診断のための出生前検査を考慮する際、医療専門家や家族の間で出生前検査の利用に関する見解の相違が存在しうる。ほとんどの施設では、個人の判断の上で出生前検査の利用を考慮するが、これらの問題について議論することは有用であろう。
関連情報
GeneReviewsスタッフは、この疾患を持つ患者および家族に役立つ以下の疾患特異的な支援団体/上部支援団体/登録を選択した。GeneReviewsは、他の組織によって提供される情報には責任をもたない。選択基準における情報については、ここをクリック。
- European Huntington's Disease Network(EHDN)
- HDBuzz
- Huntington Society of Canada
- Huntington’s Disease Africa
- Huntington's Disease Society of America(HDSA)
- International Huntington Association
- La Société Huntington du Québec(Huntington Society of Quebec)
- National Library of Medicine Genetics Home Reference
- Testing for Huntington Disease: Making an Informed Choice
- Hereditary Disease Foundation
- European Huntington's Disease Network(EHDN) Registry
- Huntington Study Group(HSG)
Germany
www.ehdn.org
151 Frederick Street
Suite 400
Kitchener Ontario N2H 2M2
Canada
Phone: 800-998-7398(toll-free); 519-749-7063
Fax: 519-749-8965
Email: info@huntingtonsociety.ca
www.huntingtonsociety.ca
Phone: 254746734559
Email: info@hd-africa.org
www.hd-africa.org
505 Eighth Avenue
Suite 902
New York NY 10018
Phone: 800-345-4372(toll-free); 212-242-1968
Fax: 212-239-3430
Email: hdsainfo@hdsa.org
www.hdsa.org
Netherlands
Email: svein@iha-huntington.org
www.huntington-disease.org
Montréal Quebec
Canada
Phone: 514-282-4272; 877-282-2444; 877-220-0226
Fax: 514-937-0082
Email: shq@huntingtonqc.org
www.huntingtonqc.org
Booklet providing information about Huntington disease and genetic testing
University of Washington Medical Center
Seattle WA
Testing for Huntington Disease: Making an Informed Choicee
3960 Broadway
6th Floor
New York NY 10032
Phone: 212-928-2121
Fax: 212-928-2172
Email: cures@hdfoundation.org
www.hdfoundation.org
Germany
EHDN Registry
95 Allens Creek
Building 1, Suite 132
Rochester NY 14618
Phone: 800-487-7671 (toll-free)
Fax: 585-672-9912
Email: info@hsglimited.org
www.huntingtonstudygroup.org
分子遺伝学
分子遺伝学とOMIMの表の情報はGeneReviewsの他の場所の情報とは異なるかもしれない。表は、より最新の情報を含むことがある。
表A. ハンチントン病:遺伝子とデータベース
| 遺伝子 | 染色体座 | タンパク | 遺伝子座特異的データベース | HGMD | ClinVar |
|---|---|---|---|---|---|
| HTT | 4p16.3 | Huntingtin | HTT database | HTT | HTT |
データは以下標準リファレンスから集約されている:遺伝子はHGNC;染色体座はOMIM;タンパクはUniProt.リンク先が貼られているデータベース(Locus Specific、HGMD、ClinVar)の説明について、ここをクリック。
表B.ハンチントン病のOMIM収録(View All in OMIM)
| 143100 | HUNTINGTON DISEASE;HD |
| 613004 | HUNTINGTIN;HTT |
分子病態
HTTはhuntingtinタンパク質をコードする。HTTのエクソン1におけるCAG三塩基リピート伸長は、huntingtinにおけるグルタミン残基の非中断伸張に翻訳される。このhuntingtinのポリグルタミン伸長は、重要な細胞プロセスを異常制御することにより毒性を発揮し、最終的に細胞死に至らせる。
特に、huntingtinは細胞骨格動態、エンドサイトーシス、小胞輸送(BDNF)、エネルギー代謝、タンパク質代謝回転、遺伝子発現(転写とRNAプロセシング)など、細胞のいくつかの不可欠な機能を高める基盤として働く。CAGリピート伸長がある場合、その役割の多くが阻害され、疾患の表現型に寄与するであろう[Saudou & Humbert 2016]。
疾患発症メカニズム. 主要なメカニズムは機能獲得であるが、多くの研究は機能消失を支持している。
表5.HTTの技術的課題
| 技術的課題 | コメント(参考文献) |
|---|---|
| リピートのシークエンシング | CAG;ただし、中断リピートである[(CAG)n-CAA-CAG]が大多数のアレルに存在する[Wright et al 2019];臨床的意義については「遺伝子型と臨床型の相関」を参照のこと。 |
| 伸長アレルの検出手法(表6参照) | 従来のPCR法、triplet-primed PCR(TP-PCR)法[Jama et al 2013]、サザンブロット[Bean & Bayrak-Toydemir 2014]が記載。 |
| 体細胞の不安定性 | 異常なCAGリピート数を有するアレルは、リピートの体細胞不安定性を示すことがある[Telenius et al 1994]。 |
| 生殖細胞の不安定性 | リピート長の伸縮は父系・母系遺伝にかかわらず起こりうるが、伸長が起こるのは父系遺伝の方がはるかに一般的で、大きな伸長(すなわち7リピートより多いCAG)は、ほぼ父系遺伝で起こる[Semaka et al 2013a]。 若年HD患者の多くは、父親から伸長したアレルを受け継いでいる[Nahhas et al 2005]。 |
HTT CAGリピートの特定方法 HTT CAG三塩基リピート伸長の検出とサイズ決定には技術的な課題があるため、CAGリピート伸長の除外または検出には複数の方法が必要となる場合がある(表6参照)。ほとんどのリピートは、従来のPCR法で検出できる可能性がある。しかし、見かけ上の正常CAGリピートのホモ接合性が検出されても、非常に大きく伸長したCAGリピートの存在を否定することはできない。したがって、triplet-primed PCR(TP-PCR)法またはサザンブロッティングによる分析が必要な場合がある。さらに、伸長したリピートの体細胞および生殖細胞における不安定性が考慮されなければならない。
表6.HTT CAGリピートの特定方法
| CAGリピート数の解釈 | 方法別に予想される結果 | ||
|---|---|---|---|
| 従来のPCR法 | Triplet-Primed PCR法1 | Expanded Repeat Analysis2 | |
| 正常:≦26 | 検出3 | 脚注1を参照 | 伸長は検出可能、リピート数は予測可能6 |
| 中間型:27-35 | 検出3, 4 | 伸長は検出される場合があるが、リピート数は特定不可能5 | |
| 病的 (低浸透性):36-39 |
検出3, 4 | ||
| 病的 (完全浸透性):≥40 |
ほとんどのアレルが検出される3, 4 | 伸長は検出されるが、リピート数は特定不可能5 | |
- Triplet-Primed PCR(TP-PCR)法は、1回のアッセイでの正常リピート数決定と伸長リピートの検出に、従来のPCR法のプライマーを含めることができる。TP-PCR法そのものは、正常範囲内のアレルであっても、リピート数を決定することはできない。
- 伸長リピートを検出し、リピート数を推測する方法には、ゲル電気泳動やサザンブロット測定によるlong-range PCR法が含まれる。リピート数の検出上限は増幅の際の正常アレルの競合により、測定デザイン、研究室、サンプル、患者毎におそらく異なってくる。
- 見かけ上のホモ接合性リピートが検出されても、CAGリピート伸長の存在を除外することはできない。したがってリピート伸長を検出するにはTP-PCR法またはexpanded repeat analysisによる分析が必要である。
- PCR法を基盤とした方法は、115CAGリピートまでのアレルを検出する[Potter et al 2004, Levin et al 2006]。他の方法が、時に大きなCAGリピートを同定するのに有用な場合がある。リピート数の検出上限は増幅の際の正常アレルの競合により、測定デザイン、研究室、サンプル、患者毎におそらく異なってくる。
- この範囲の下限のリピートは、伸長アレルを示唆する特徴的なスタッターパターンを示さない場合がある。
- CAGリピート伸長に対するサザンブロットは、以下記載[Bean & Bayrak-Toydemir 2014]。
表7.重要なHTTバリアント
| 参照シーケンス | DNA塩基変化 | 予想されるタンパク質変化 | リピート範囲 |
|---|---|---|---|
| NM_002111.8 NP_002102.4 |
c.52_54CAG[9_26] | p.Gln18[9_26] | 正常 |
| c.52CAG[27_35] | p.Gln18[27_35] | 中間型 | |
| c.52CAG[36_39] | p.Gln18[36_39] | 低浸透性 | |
| c.52CAG[40_?] | p.Gln18[40_?] | 完全浸透性 |
表中のバリアントは、著者らにより提供。GeneReviewsスタッフは、独自にバリアント分類を検証していない。
GeneReviewsはHuman Genome Variation Society(varnomen.hgvs.org)による標準記名法にならう。記名法に関する解説は、Quick Referenceを参照のこと。
更新履歴:
- GeneReview 著者 :Simon C Warby, PhD, Rona K Graham, PhD, Michael R Hayden, MB, ChB, PhD, FRCP(C), FRSC.
日本語訳者: 窪田美穂(ボランティア翻訳者),櫻井晃洋(信州大学医学部附属病院 遺伝子診療部)
Gene Review 最終更新日: 2010.4.22. 日本語訳最終更新日: 2010.6.16. - Gene Reviews著者: Simon C Warby, PhD, Rona K Graham, PhD, and Michael R Hayden, MB, ChB, PhD, FRCP(C), FRSC.
日本語訳者: 和泉 賢一 (札幌医科大学医学部遺伝医学,NGSDプロジェクト)
Gene Reviews 最終更新日: 2014.12.11 日本語訳最終更新日: 2018.3.16 - Gene Reviews著者: Nicholas S Caron, PhD, Galen EB Wright, PhD, and Michael R Hayden, MB, ChB, PhD, FRCP(C), FRSC
日本語訳者: 冨成麻帆(名古屋市立大学大学院看護学研究科)、佐橋健太郎(名古屋大学医学部附属病院 脳神経内科)、勝野雅央(名古屋大学大学院医学系研究科 神経内科学)
GeneReviews最終更新日: 2020. 6.11. 日本語翻訳最終更新日:2022.10.30.[in present]
![]()