ムコ多糖症ⅣA型
(Mucopolysaccharidosis TypeⅣA)
[同義語:MPSⅣA, Morquio A Disease, Morquio Syndrome Type A]
Gene Reviews著者: Debra S Regier, MD, PhD, Matthew Oetgen, MD, and Pranoot Tanpaiboon, MD.
日本語訳者: 和田宏来(県西総合病院小児科/筑波大学大学院小児科)
Gene Reviews 最終更新日: 2016.3.24.日本語訳最終更新日:2017.6.29.
原文 Mucopolysaccharidosis TypeⅣA
要約
疾患の特徴
ムコ多糖症ⅣA型(MPSⅣA)の臨床型スペクトラムは、重症で急速に進行する早期発症型から緩徐に進行する後期発症型まで幅広く連続している。ムコ多糖症ⅣA型患児は、出生時にははっきりとした臨床所見を認めない。重症型は通常1-3歳の間に明らかになってくるが、脊柱後側弯症、X脚(外反膝)、鳩胸がしばしば初発症状となる。緩徐進行型は小児期後期/思春期まで明らかにならないことがあり、初発症状はしばしば臀部の問題(疼痛、こわばり、レッグ・ペルテス病)である。進行性の骨・関節病変のため低身長となり、やがて動けないほどの痛みや関節炎をきたす。他の臓器系統の病変により、呼吸器合併症、睡眠時無呼吸、心臓弁疾患、聴覚障害、角膜混濁による視覚障害、歯の異常、肝腫大など、重大な障害が引き起こされる。脊髄の圧迫はよく認められる合併症で、神経障害をきたす。発症初期における患児の知能は正常である。
診断・検査
ムコ多糖症ⅣA型の診断は、N-アセチルガラクトサミン-6-スルファターゼ(N-acetylgalactosamine 6-sulfatase, GALNS)酵素活性の測定、もしくは分子遺伝学的検査でGALNS遺伝子の両アレル病原性変異を同定することによりなされる。
臨床的マネジメント
症候の治療:
酵素補充療法(エロスルファーゼアルファもしくはビミジム®)が利用可能であるが、ムコ多糖症ⅣA型の骨格異常やそれ以外の異常に対する長期的な効果については不明である。ムコ多糖症ⅣA型患者の評価・治療は、複雑な医学的問題を抱える患者の診療を得意とする医師と協力しながら、複数の専門家が協同して行うのが最もよい。物理医学専門医(physiatrists)、理学療法士、作業療法士は可動性や自律性の最適化を補助する。心理的サポートにより、対処能力やQOLを最適化することができる。教職者は医学的に虚弱な患者の学習環境を最適化することができる。下肢のアライメント異常、股関節亜脱臼や股関節痛、上位頸椎不安定症、進行性の脊柱後弯症に対してしばしば外科手術が必要となる。上肢の治療に、外側の手関節固定装具もしくは部分/全手関節固定術などを行うことがある。心臓弁疾患に対して生体弁/人工弁置換術を必要することがある。上気道閉塞や閉塞性睡眠時無呼吸では、肥大した扁桃/アデノイドを切除する。びまん性の気道狭窄に対して、陽圧換気や気管切開を必要とすることがある。角膜混濁に対する角膜形成術の転帰はさまざまである。難聴に対して、はじめは換気チューブ、後に補聴器によって治療することが多い。
二次合併症の予防:
脊椎奇形に続発する麻酔の術前/術後合併症や気道管理の難しさを予測しておく必要がある。すべての患者は、定期接種と同様にインフルエンザや肺炎球菌の予防接種を受けるべきである。人工弁置換術を受けた患者、心臓弁修復術で人工物を使用した患者、感染性心内膜炎の既往がある患者では感染性心内膜炎の予防が推奨される。
定期検査:
酵素補充療法を受ける患者:少なくとも6ヶ月ごとに身体診察を行う。QOLの指標や肺機能を年1回評価する。
すべての患者:心臓血管系、肺、筋骨格系、神経系の機能を評価するために年1回負荷試験をおこなう。上下肢の機能やアライメント異常、股関節の形成不全や亜脱臼、胸腰椎の後弯症を認めないか年1回評価する。脊髄圧迫を評価するため6ヶ月ごとに神経学的診察と頸椎X線を行う。1~3年ごとに全脊椎MRIを行い屈曲・伸展像を撮影する。心拍数の評価と心電図を年1回、疾患の経過により心エコーを1~3年ごとに施行する。閉塞性睡眠時無呼吸に対する評価や肺機能検査を年1回行う。ムコ多糖症ⅣA型患者用の成長曲線を用いて栄養状態を評価する。外来受診時に毎回視力検査や眼科検査、歯科診察を6~12ヶ月ごと、聴力検査(オージオグラム)を年1回施行する。
避けるべき薬物/環境:
過剰な体重増加、β遮断薬は避けるべきである。
遺伝カウンセリング
ムコ多糖症ⅣA型は常染色体劣性遺伝性疾患である。受胎時に罹患者の同胞が罹患している確率は25%、無症候性キャリアである確率は50%、罹患もしておらずキャリアでもない確率は25%である。リスクのある家族に対する保因者診断やリスク妊娠における出生前診断は、家系での病原性変異が同定されている場合可能である。
診断
示唆される所見
病歴、身体診察、骨格系X線、眼科診察で以下のような所見を認めた場合にムコ多糖症ⅣA型を疑うべきである。
病歴
- 出生時にははっきりとした臨床所見を認めない。
- アデノイド摘出術、扁桃摘出術、ヘルニア修復術、鼓膜換気チューブ(すべてのムコ多糖症に一般的な所見)の既往がある。
- 頸椎圧迫/癒合の既往歴、もしくは四肢のアライメントに関する手術歴(すべてのムコ多糖症の中でⅣA型に特有)がある。
- 呼吸器合併症(睡眠時無呼吸・耐容能の低下・いびき)を認める。
- 心臓弁疾患を認める。
- 歯の異常を認める。
身体診察
重症のムコ多糖症ⅣA型では、通常以下の所見を1~3歳の間に認める。緩徐進行型では、以下の所見は10代になるまで明らかにならないことがある。
- かなり均整のとれていない低身長で、体幹は短く四肢は正常である(指端距離は身長を上回る)。
- 手関節の尺側偏位(図1および図2)
- 鳩胸と胸郭下部の拡大(図3および図4)
- 亀背(短い部分の構造的な胸腰椎後弯症により背部が鋭角となる)、脊柱後弯症、脊柱側弯症
- X脚(外反膝)(図5)
- 過可動関節
- 頻繁な転倒を伴う動揺歩行
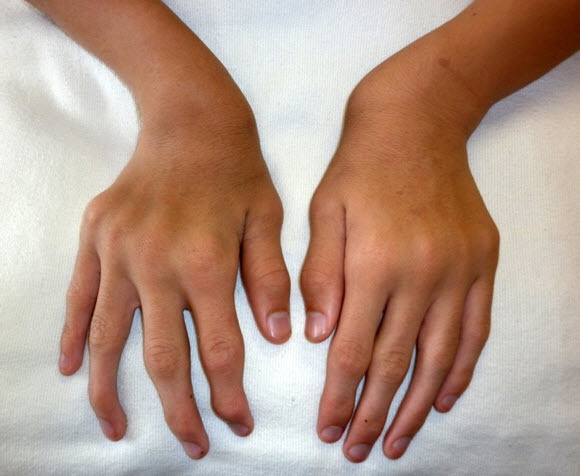
図1 15歳男性、手関節が腫大しており尺側に偏位している
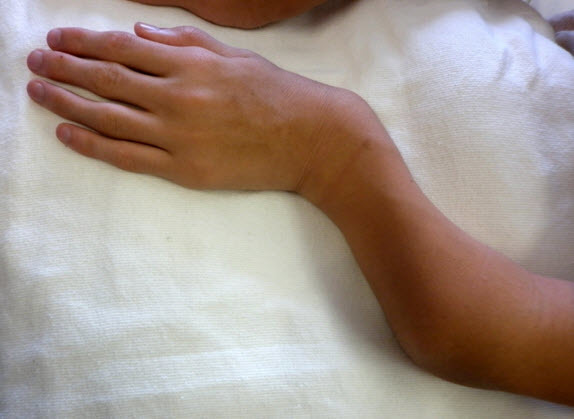
図2 15歳男性、短い前腕と手関節の尺側偏位を認める
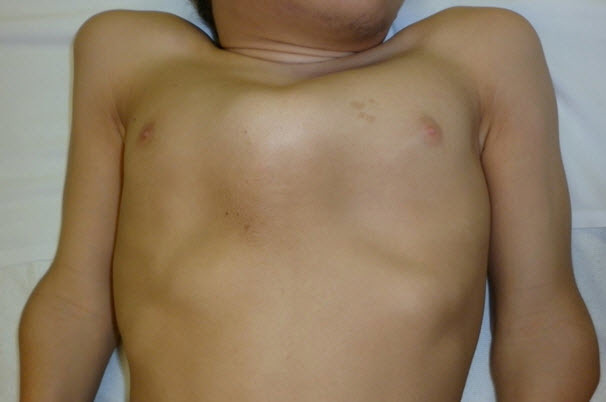
図3 15歳男性、胸部の異常および短い頸部を認める
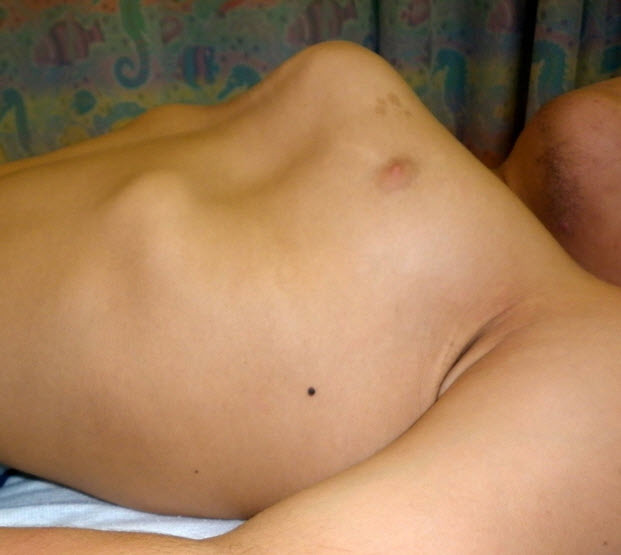
図4 15歳男性、側面像で重度の鳩胸を認める
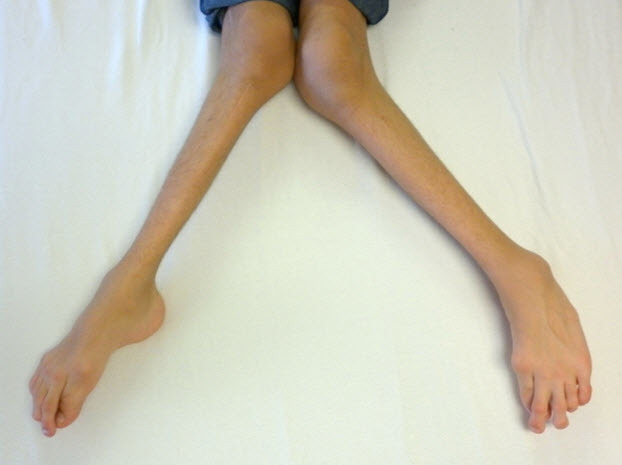
図5 15歳男性、重度のX脚(外反膝)を認める
骨格系X線
- 歯突起形成不全とそれに続発する頸椎不安定症(図6)
- 脊柱後弯症(脊椎がカーブ状になって背部の弯曲/円形化をきたし、脊柱後弯症/前傾姿勢に至る)(図7)
- 1つ以上の隣接する椎体の楔状化を伴った亀背(構造的な脊柱後弯症)(図7)
注:急速進行型では出生時にX線で腰椎の異常を認めうる。 - 脊柱側弯症
- 鳩胸もしくは(頻度は低いが)漏斗胸
- 短い尺骨、橈骨骨端の尺側偏位、骨成熟の遅延
- 中手骨は短く、第2~第5中手骨の遠位端が円型もしくは尖っている
- 拡大した腸骨翼、大腿骨骨端の平坦化(図8)、外反股
注:骨格系の異常は身体的異常より前に認められる。
眼科診察
角膜混濁、乱視、網膜症による視力障害を認める。
示唆される検査所見
- 定性尿中グリコサミノグリカン分析は、薄膜クロマトグラフィーもしくは電気泳動を用いてケラタン硫酸やコンドロイチン-6-硫酸のような特異的なグリコサミノグリカンを検出する方法である。
注:尿中グリコサミノグリカン量の異常を伴って、もしくは伴わずに、一部の患者に(定性分析で)ケラタン硫酸が認められている。
- 定量尿中グリコサミノグリカン分析ではグリコサミノグリカンの総量を測定する。
・ケラタン硫酸の上昇は、N-アセチルガラクトサミン-6-スルファターゼ(ⅣA型)、B-ガラクトシダーゼ(ⅣB型)いずれかの欠損を示唆する。
注:高齢者では軟骨形成が減少しているため、18歳以下の若年患者における尿中ケラタン硫酸値は高齢者よりも高い。 - コンドロイチン-6-硫酸の上昇は、N-アセチルガラクトサミン-6-スルファターゼ(ⅣA型)の欠損を示唆する。
- 尿中ケラタン硫酸分析は、標準的な色素ベースの総グリコサミノグリカン定量分析よりも感度が高く、将来的に尿中グリコサミノグリカン定量に代わる検査となる可能性がある。
- 一部の患者では、定性・定量尿中グリコサミノグリカンはともに正常となりうる。そのため、ムコ多糖症Ⅳ型の臨床症候を認める小児では、たとえグリコサミノグリカン分析が正常であっても、さらに酵素や分子学的な評価を行う必要がある。
診断の確定
発端者におけるムコ多糖症ⅣA型の診断は、(1)培養線維芽細胞もしくは白血球でN-アセチルガラクトサミン-6-スルファターゼ(GALNS)酵素活性が低い、もしくは(2)分子遺伝学的検査でGALNS遺伝子に両アレル変異を認めることで確定する(表1を参照)。
分子検査には単一遺伝子検査もしくは多遺伝子パネルがある。
- 単一遺伝子検査 GALNS遺伝子のシークエンス解析がまず行われ、病原性変異が1つしか見つからない/もしくは認めない場合はつづけて標的遺伝子の欠失/重複解析を行う。
- GALNS遺伝子および他の関連遺伝子(「鑑別診断」の項を参照)を含む多遺伝子パネルも考慮することがある。
注:(1)パネルに含まれる遺伝子やそれぞれの遺伝子に用いられる検査の感度は検査機関や時期によって異なる。(2)一部の多遺伝子パネルには、このGeneReviewで触れていない病態と関連する遺伝子も含まれている可能性がある。そのため、臨床医は多遺伝子パネルがもっとも合理的なコストでその病態の遺伝的な原因を追究できるかどうかを見極める必要がある。
分子遺伝学的検査
IDS遺伝子変異の同定により、男性発端者におけるムコ多糖症Ⅱ型の診断が確定する。また、IDS遺伝子変異の同定は、臨床型が非典型的な場合もしくはグリコサミノグリカン分析結果と臨床型が合わない場合に有用なことがある。
ムコ多糖症Ⅱ型患者のIDS遺伝子変異には大きく3つのタイプがある。
- 遺伝子内の病原性変異(82%)
- 1つ以上のエクソンもしくは遺伝子全体の欠失(9%)
- 主にIDS遺伝子の25kbテロメア側に存在している偽遺伝子IDSP1との組み換えにより生じる複雑な遺伝子再構成(9%)
表1ムコ多糖症ⅣA型で用いられる分子遺伝学的検査
| 遺伝子1 | 検査方法 | 検査方法によって同定された変異2 を有する発端者の頻度 |
|---|---|---|
| GALNS | シークエンス解析3 | 94%4 |
| 標的遺伝子の欠失/重複解析5 | 2%-3%6,7 |
- 染色体座位と蛋白については、表A「遺伝子・データベース」を参照。
- この遺伝子で同定されたアレル変異に関する情報については、「分子遺伝学」の項を参照。
- シークエンス解析では、良性の変異、良性と考えられる変異、臨床的意義が不明の変異、病原性と考えられる変異、病原性変異が検出される。病原性変異には、小さな遺伝子内欠失・挿入、ミスセンス変異、ナンセンス変異、スプライス部位変異が含まれるが、典型的にはエクソンや遺伝子全体の欠失/重複は検出できない。シークエンス解析の結果の解釈について考慮すべき問題はこちらをクリック。
- Caciottiら(2015)。遺伝子変化の多くはヌクレオチド1-2個の増減であるが、イントロン変化はその9%を占める。
- 標的遺伝子の欠失/重複解析では遺伝子内欠失/重複を同定する。用いられる方法には、定量PCR、ロングレンジPCR、MLPA(multiplex ligation-dependent probe amplification)法、単一エクソンの欠失/重複を検出する標的染色体マイクロアレイ解析などがある。
- 病原性変異の約2.7%は大きな欠失による。
- 1例は16番染色体のテロメア末端の母性ダイソミーを有して発症、2例はGALNS遺伝子のエクソン10-14およびエクソン9-14を含む大きな欠失を有していた。これは染色体マイクロアレイ法(chromosomal microarray, CMA)によって同定されることがある。
以下の場合にGALNS酵素活性を用いることができる。
- 臨床所見はムコ多糖症ⅣA型を強く示唆するが、尿中グリコサミノグリカン分析は正常の場合。
かつ/もしくは - 分子遺伝学的検査でGALNS遺伝子に両アレル病原性変異を同定できない場合。
くわえて、酵素解析によるムコ多糖症ⅣA型の確定診断は、意義不明のシークエンス変異の解釈に役立つ可能性がある。GALNS酵素活性は培養線維芽細胞/白血球で測定できる。各検査機関で酵素活性の正常範囲は異なるため、検査機関が異なる場合は結果を直接比較することはできない。残存酵素活性の値は疾患の重症度と相関する可能性がある。
注:
- 酵素活性の低下は以下のような他疾患でも認めうる。
- 多種スルファターゼ欠損症(multiple sulfatase deficiency, MSD)、すなわち(GALNSを含む)複数のスルファターゼ活性の欠損。そのため、GALNS酵素活性に異常を認めた場合、くわえてMSDの評価のためスルファターゼ酵素を測定する必要がある。
- ムコリピドーシスⅡ型およびムコリピドーシスⅢ型(ムコリピドーシスⅢ型α/β、ムコリピドーシスⅢ型γを参照)。ムコリピドーシスⅡ型およびムコリピドーシスⅢ型では、ライソゾーム酵素を標的とするマンノース6-リン酸は阻害され、線維芽細胞においてライソゾーム酵素活性は低下、血漿では酵素活性は上昇するが、白血球では相対的に変化は認められない。
- ムコ多糖症ⅣA型とⅣB型の臨床症状は区別できないため、慣習的に同時にB-ガラクトシダーゼ酵素活性を測定する。
臨床的特徴
臨床像
ムコ多糖症ⅣA型の臨床像は、重症で急速に進行する早期発症型から緩徐に進行する後期発症型まで幅広く連続している。過去には、この両者は身長、骨の変形による主観的な重症度評価、生存率から区別していた。しかし、臨床所見および生化学所見のいずれによっても2つの病型を明確に区別することはできない。そのため、ムコ多糖症ⅣA型の臨床型は重症から緩徐進行型まで連続的であると考えるべきである。
患児は出生時には明らかな臨床所見を認めない。重症型は通常1-3歳の間に顕在化してくる。緩徐進行型は小児期後期もしくは思春期まで明らかとならない可能性がある。
両病型とも、初発症状はさまざまで1つのことも複数のこともある。脊柱後側弯症、X脚(外反膝)(図5)、鳩胸(図3および図4)が重症型でもっとも多い初発症状である。対して、(大腿骨近位部の圧壊や平坦化による)疼痛や硬直を含む股関節の問題は緩徐進行型で多い初発症状である。
過去に報告されたムコ多糖症ⅣA型の自然歴はⅣA型(モルキオ症候群A型、患者の95%以上を占める)とⅣB型(モルキオ症候群B型、患者の5%未満)を区別していないため、以下の情報はⅣA型・ⅣB型両方に関連する。
骨格系の異常はⅣA型の代表的な特徴であるが、他の臓器系統の病変により、呼吸器合併症・閉塞性睡眠時無呼吸・心臓弁疾患・聴力障害・角膜混濁・歯の異常・肝腫大といった重大な症状をきたしうる。とくに後年になって疾患に気付かれた場合は脊髄の圧迫により神経障害を呈している。
粗な顔貌も認められるが他のムコ多糖症よりも軽度である。
ムコ多糖症ⅣA型患児の知能は典型的には正常である。
靭帯弛緩や過可動関節はムコ多糖症ⅣA型に特有の徴候であり、他の蓄積病ではまれである。
筋骨格系
骨格系の異常は経過とともに悪化する。骨・関節病変は疼痛や関節炎を起こし、ひいては身体障害に至る。
上肢病変も進行性であり、手-手関節の筋力が低下し、フォークを使うなど日常生活の活動が一部制限されうる。手関節の過可動と尺側偏位はムコ多糖症ⅣA型に特有の徴候である。
下肢病変は広く認められ、未治療の場合進行性であり、一般的には進行性の股関節亜脱臼によるアライメント不整および外反変形を呈する。それにより歩行に著しい変化を認めうる。活動により臀部・膝・足関節が痛む。持久力が低下する。
膝および足関節の外反はもっともよく認められる下肢の変形である。X脚(外反膝)は大腿骨遠位部と脛骨近位部の病変や関節の弛緩による。
PEDI(Pediatric Evaluation of Disability Inventory)やFIM(Functional Independence Measure)を用いた縦断研究では、ムコ多糖症Ⅳ型患者の関節可動域は大きく制限され、一般的に晩期には歩行不能となることが示された。可動域の最適化のためには、理学療法士およびリハビリの専門家から構成されるチームによる積極的かつ長期的な介入がしばしば必要となる(「臨床的マネジメント」を参照)。
股関節形成異常 疾患早期では、大腿骨頭は小さく寛骨臼は浅い。大腿骨頭および寛骨臼の破壊が進行して股関節脱臼・関節炎・重度の関節可動域制限をきたし、歩行不能となる。
脊髄圧迫はどの脊髄分節でも起こることがある。頸髄圧迫がもっともよく認められる。頸椎不安定症、異常な歯突起に合併した非石灰化線維軟骨、靭帯弛緩、環軸椎関節における軟骨・靭帯の肥大、硬膜外腔へのグリコサミノグリカン蓄積、椎間板突出、胸腰椎後弯症、後天性の脊柱管中心部狭窄によっても脊髄圧迫は起こりうる。
環軸椎不安定症をきたす歯突起形成不全は患者の90%に認められ、のちに上位頸髄の圧迫を起こすことがある(図6)。
大したことのない転落や頸部進展で四肢不全麻痺/突然死をきたしうるため、環軸椎不安定症を有する場合、未治療ではしばしば10-20代を超えて生存できない。
脊柱管狭窄はびまん性もしくは限局性である。脊柱管狭窄症の原因は脊髄圧迫の原因と類似しており、脊柱後弯症、椎間板突出、後縦靭帯の全体的な肥厚、グリコサミノグリカン蓄積による椎間靭帯の肥厚などがある。
年長患者では腰椎アライメント不整(すなわち胸腰椎後弯症)により限局性の脊柱管狭窄症、圧迫性脊髄症、対麻痺に至ることがある。
靭帯弛緩による関節過可動はよく認められる。しかし、関節可動域の減少も膝・股・肘といった大関節で見られうる。
神経系
診断時、典型的にはムコ多糖症Ⅳ型(すなわちⅣA型およびⅣB型)患者の発達は正常で、知能も正常である。ムコ多糖症Ⅳ型の神経学的所見は、ほとんどの場合頸髄や腰髄の異常に続発する。神経学的合併症のリスク上昇により、ムコ多糖症ⅣA型患児は非罹患者よりも発達遅滞や学習障害が多く認められる。
血管周囲腔が目立つ、側脳室の拡大、前頭部の脳脊髄液が目立つといった些細な脳MRI異常がムコ多糖症ⅣA型患者14人のうち8人で認められたと報告されている。同じ研究では認知機能を評価しており、不安・抑うつ・注意持続時間の減少・身体的愁訴を含む行動上の問題が明らかとなった。これらの所見が、疾患に特異的な生化学的異常によるのか、もしくは慢性疾患によるのか、それともその2つの組み合わせによって起こるのかは不明である。
循環器
循環器合併症には、心室肥大、早期発症で重症の弁疾患などがある。冠動脈内膜硬化も報告されている。
ムコ多糖症ⅣA型患者325人を対象とした多施設・多国籍の横断研究(MorCAP)では、弁逆流症は弁狭窄症よりも多く認められた。弁逆流症のなかで、三尖弁逆流症が最も多かった(35%)。僧帽弁逆流症、大動脈弁逆流症、肺動脈弁逆流症がそれぞれ25%、19%、14%に認められた。ムコ多糖症ⅣA型患者は通常、小さな左室径と少ない心拍出量を代償するため心拍数の増加や心拍出係数の上昇がみられる。
呼吸器
呼吸器合併症は重大な症状/死因の主たる原因である。気道閉塞、睡眠時呼吸障害、拘束性肺疾患が報告されている。アデノイド(咽頭扁桃)、口蓋扁桃、咽頭、喉頭、気管、気管支樹(bronchial tree)へのグリコサミノグリカン蓄積により、扁桃肥大、気管の歪み(tracheal distortion)、気管/気管支軟化症、閉塞性睡眠時無呼吸を呈する。気管や気管支へのグリコサミノグリカン沈着により気道が歪み、弯曲や頸部屈曲時に気道閉塞をきたすこともありうる。(とくに頸椎融合/安定化手術を施行中に)気付かれていない場合や屈曲位で頭頸部が癒合している場合、急な閉塞で抜管に至ることがある。
拘束性肺疾患は、小さな胸郭、胸壁異常、脊椎変形、頸髄障害、肝腫大による横隔膜挙上などにより起こる。
環軸椎不安定症や上気道閉塞のため、頸部の伸展を保ち気道の歪みを最小限にするように、ムコ多糖症ⅣA型患者は平坦な場所では枕をつかわず腹臥位を好む。
呼吸器合併症に気付かれていないか未治療の場合、肺性心や呼吸不全をきたし、早期に死亡する。
成長
ムコ多糖症ⅣA型患児の出生時体重は正常で、身長はより高い。
1~3歳の間に非罹患者に比べて成長速度は低下する。
18歳における平均身長は、非罹患者では男性177cm、女性163cmであるのに対し、男性123cm、女性117cmである。
眼
ムコ多糖症ⅣA型患者の50%以上に眼科的所見が認められる。自然経過の研究は行われていない。そのため、眼科的所見の発症年齢を予測することはできない。
もっとも多く認められるのは、患者(1-65歳)の50%に認められる緩徐に進行する角膜混濁である。
より少ない他の眼科的所見には、乱視、白内障、点状水晶体混濁、開放隅角緑内障、視神経乳頭腫脹、視神経萎縮、網膜症がある。眼窩が浅いため眼が突出する偽眼球突出は兎眼性角膜炎を起こし、外見上の問題にもなりうる。
歯
乳歯の萌出は正常で、間隔は広く、薄い不整な(小さな斑点で覆われた)エナメル質で変色しており、小さな突起のある歯尖は正常な摩耗で次第に平らとなる。
永久歯もエナメル質の形成不全が認められる。
聴覚
ムコ多糖症ⅣA型では軽度~中等度の難聴はよく認められる。聴力障害はしばしば10歳になる頃に認められる。
混合性難聴(すなわち伝音性および感音性の両方)は、伝音性、感音性単独よりも多くみられる。
伝音性難聴は反復する中耳の感染症、滲出性中耳炎、耳小骨の変形などによる。
内耳もしくは中枢神経系へのグリコサミノグリカン蓄積による感音性難聴が報告されている。
遺伝子型と臨床型の関連
欠失やノンセンス変異のような、蛋白機能を重度に障害すると予測されるGALNS遺伝子の変化は、成長障害が急速に進行する患者でよく認められる。
対照的に、p.Thr312Serのような保存的な変化を有する患者の臨床型はより軽症の傾向にあり、酵素活性が部分的に保たれているためである可能性が高い。
c.898+1G>Cのホモ接合を有する患者は緩徐に進行する経過をとる。
命名
ムコ多糖症ⅣA型(N-アセチルガラクトサミン-6-スルファターゼ欠損もしくはGALNS遺伝子の両アレル病原性変異)やムコ多糖症ⅣB型(B-ガラクトシダーゼ欠損もしくはGLB1遺伝子の両アレル病原性変異)の基礎が理解されるより以前から、古くからたくさんの研究が行われ、より詳細に自然歴が記述されてきた。そのため、ムコ多糖症Ⅳ型という病名は、酵素や分子学的診断によらず臨床診断に基づく。その一方で、より特異的なムコ多糖症ⅣA型やⅣB型という病名は酵素/分子学的診断による。
モルキオ症候群としても知られるムコ多糖症ⅣA型は、Morquio(1929)とBrailsford(1929)によって初めて特徴がまとめられた。
ムコ多糖症ⅣA型およびⅣB型は、それぞれモルキオ症候群A型およびB型として知られている。
発生率
ムコ多糖症ⅣA型はまれである。オーストラリアでの発生率は926,000人に1人と推定されている。一方、英国では599,000人に1人と推定されている。複数の国における出生有病率は71,000~599,000人に1人である。ドイツからの報告によると、ムコ多糖症ⅣA型の発生率は270,000人に1人、ⅣB型は1,000,000人に1人未満である。同様に、イタリアにおけるムコ多糖症ⅣA型の発生率は、出生300,000人に1人と推定されている。
遺伝学的関連(アレル)疾患
このGeneReviewで述べている他にGALNS遺伝子変異と相関する臨床型はない。
鑑別診断
ムコ多糖症ⅣB型 もっともムコ多糖症ⅣA型に類似しているのはムコ多糖症ⅣB型であり、B-ガラクトシダーゼ酵素をコードするGLB1遺伝子の両アレル変異によりケラタン硫酸の蓄積が起こる。ムコ多糖症Ⅳ型の臨床所見を呈するほとんどの者において、ⅣA型とⅣB型の鑑別は生化学検査や分子遺伝学的検査によってのみ行える。
GLB1遺伝子の両アレル変異は、神経学的予後が悪く骨系統疾患を伴うライソゾーム病であるGM1ガングリオシドーシスも起こす。GM1ガングリオシドーシスの臨床像は、乳児型、乳児後期型、若年型、成人型と連続している。乳児型ではしばしば2歳までに死亡するが、成人型では生命予後は悪くないことがある。
ムコ多糖症ⅣB型で同定される新規GLB1変異はしばしばその蛋白の基質結合領域に位置するが、一部の変異はGM1ガングリオシドーシスとムコ多糖症ⅣB型の双方に関連する。
その他のムコ多糖症 ムコ多糖症ⅣA型の徴候や症状は、広範囲の臨床所見を呈する他のムコ多糖症と重複している。ムコ多糖症Ⅰ型、ムコ多糖症Ⅱ型を参照。
他のムコ多糖症と比べて、ムコ多糖症ⅣA型では知能は正常で粗な顔貌が目立たないことが特徴である。一般的に、ムコ多糖症ⅣA患者の視力は他のムコ多糖症患者よりも良好である。くわえて、過可動関節はムコ多糖症Ⅳ型に特有である。環軸椎不安定症は他のムコ多糖症よりⅣ型で多く認められる。
OMIMでこの臨床型に関連する遺伝子を閲覧するにはムコ多糖症:OMIM臨床型シリーズを参照のこと。
脊椎骨端異形成症は同様のX線所見を呈する。臨床徴候、とくに骨外性の特徴により脊椎骨端異形成症とムコ多糖症ⅣA型を鑑別できる可能性がある。
シムケ免疫性骨形成不全およびX連鎖性遅発性脊椎骨端異形成症を参照。
レッグ・カルベ・ペルテス病(OMIM) ときに軽症のムコ多糖症ⅣA型は発症時に股関節痛のみしか呈せず、初期にはレッグ・カルベ・ペルテス病と間違われることがある。
臨床的マネジメント
初期診断後の評価
ムコ多糖症ⅣA型と診断された患者の疾患の広がりやニーズを把握するために、以下の評価が推奨される。
- ベースラインの神経学的診察で脊髄圧迫の徴候を評価する。
- 単純X線
- 前後像・側面像・屈曲伸展位をふくむベースラインの頸椎X線。
- 全脊椎のベースラインの前後像および側面像。
- 骨盤のベースラインの前後像および蛙肢位側面像。
- ベースラインの下肢の立位前後像。1人で立つことができ、下肢の不整合の徴候がある患者では、股関節から足関節まで下肢アライメントのベースラインの立位像。
- MRI
- 潜在的な脊髄圧迫(後頭頸部・頸胸部・胸腰部)に焦点をあてた全脊椎(中立位)のベースラインMRI
- 臨床的に適応がある、たとえば単純X線で頸椎不安定症が疑われる場合、屈曲伸展位の頸椎MRI
?脳MRIは通常鎮静下や麻酔下に施行されるため、上位頸椎不安定症を評価する頸椎X線は麻酔をかける前に行うべきである。- 単純X線で頸椎不安定症が疑われる、もしくは確定的ではない所見を認めた場合、屈曲伸展位の頸椎MRIで脊髄圧迫の評価を行うことができる。
- マッケンジーら(2013)は、小児骨系統疾患患者における鎮静/麻酔下の屈曲伸展位頸椎MRIは十分な監督下であれば安全で、その結果は外科手術の適応決定に有用であると報告した。
- 以下による評価
- 物理医学専門医(すなわち理学療法やリハビリテーションの専門家)による可動性や自律性の評価。歩行制限のある患者では、心血管系、呼吸器、筋骨格系、神経系の機能評価に、6分間歩行検査や25フィート歩行時間測定をふくむ耐容能検査が用いられてきた。6分間歩行検査および25フィート歩行時間測定は診断時に行うべきである。
- 理学療法士による関節の動きの範囲や可動性の評価。
- 作業療法士による巧緻運動機能検査のような日常生活の活動性の評価。握力計による握力やピンチメーターによるピンチ力を含めて、ベースラインの上肢筋力評価も行うべきである。
- 臨床遺伝専門医や遺伝カウンセラーへの診療依頼
- 心電図および心エコー検査。冠動脈病変が疑われる場合、非侵襲的な画像検査を行うべきである。
- 呼吸内科医によるベースラインの肺機能検査およびポリソムノグラフィー
- 聴力の評価
- 歯の評価
- 視力や眼圧など眼科的評価
- ムコ多糖症ⅣA型用成長曲線への身長・体重の記入
- 疼痛の評価やQOLに関する質問票
- 物理医学専門医(すなわち理学療法やリハビリテーションの専門家)による可動性や自律性の評価。歩行制限のある患者では、心血管系、呼吸器、筋骨格系、神経系の機能評価に、6分間歩行検査や25フィート歩行時間測定をふくむ耐容能検査が用いられてきた。6分間歩行検査および25フィート歩行時間測定は診断時に行うべきである。
病変に対する治療
ムコ多糖症ⅣA型患者の治療は、複雑な医学的問題を抱える患者の診療を得意とする医師と協力しながら、以下のような複数の専門家が協同して行うのが最もよい。
- 物理医学専門医(可動性や自律性を最適化する理学療法やリハビリテーションの専門家)
- 可動性を最適化する理学療法士
- 自律性を最適化する作業療法士
- 対処能力(coping skills)やQOLを最適化する心理的サポート
- 医学的に虚弱な患者の学習を最適化する教職者
- 患者、両親、同胞、その他の家族に対し、経験を日常化するのに有用な家族療法の紹介を考慮する。
- 複数の医療機器が必要な患者に対する在宅医療
- 終末期におけるホスピス
酵素補充療法
組み換えヒトGALNS酵素補充療法(エロスルファーゼアルファもしくはビミジム®)が2014年2月に食品医薬品局(FDA)によって承認された。
- 推奨量は2mg/kg/週の静注である。酵素補充療法は根治的ではないが、運動耐容能や全体的なQOLを改善しうる。
- 投与時反応の予防のために、(可能であれば)非鎮静性抗ヒスタミン薬の前投与(各投与の30-60分前)が推奨される。解熱剤を併用することもある。
- 第Ⅲ相試験において、2mg/kg/週の投与群は対照群と比べて6分間歩行距離に統計学的に有意な改善が認められた。3分間階段昇降検査や呼吸機能検査は治療によって改善したが、統計学的に有意な差は認められなかった。
- ムコ多糖症ⅣA型の骨系統疾患に対するこの治療法の長期的な効果についてはまだ不明である(「研究中の治療法」を参照)。酵素は無血管組織にはあまり分布しないため、筋骨格系の病理に対する酵素補充療法の効果は限定的である可能性がある。
筋骨格系
発行されている整形外科的マネジメントのガイドラインについては、Whiteら(2014)の文献を参考にされたい。身体活動性のレベルは、関節の損傷やアライメント不整、脊髄損傷を予防しながら可動性を最適化するため、整形外科学、神経学、物理医学、理学療法の各専門家によってモニターされるべきである。
上肢 外側の手関節固定装具のような非侵襲的な治療を考慮することがある。手関節を安定化させるため、部分/全手関節固定術のような外科手術が必要になることがある。
膝や足関節の外反 下肢のアライメント不整で、力学的に望ましくないアライメントや運動耐容能の低下が進行する場合は治療が必要となる。しかし絶対的な適応は存在しない。
- guided growthとも呼ばれる成長調節(一次的に成長板を外科的に連結して緩徐に変形を修復する)やアライメント修正骨切り術(realignment osteotomies)の成功が報告されている。
- 早期の発見・評価により、骨端線(成長板)閉鎖前の小児患者における軽度~中等度の下肢角状変形に対して、外科的な成長板の連結を行うことができる可能性がある。典型的にはこの手技の侵襲は少なく、アライメント修正骨切り術より早期に回復する。
- 成長板が閉鎖してしまうと、急速/緩徐(外固定具を用いる)な下肢アライメント不整の治療のためには、大腿骨遠位部や脛骨近位部の骨切り術が必要となる。
- 足関節のアライメント不整は、しばしば脛骨遠位部の骨切り術(a distal tibial osteotomy)およびスクリューによる脛骨遠位部の半骨端閉鎖(distal tibial screw hemiepiphyseodesis)によって治療される。
股関節形成異常 外科手術により疼痛やアライメント不整を治療し、適切な可動性を得ることができる。
- 股関節再建術として、軽症例には大腿骨/寛骨臼いずれかの骨切り術、重症例には寛骨臼・大腿骨の骨切り術を施行する。寛骨臼が浅いため、寛骨臼骨の補強、および腸骨内板からの皮質骨移植片を用いたカスタマイズされたインプラントが通常必要である。
・若年成人で再建術では回復しない著しい股関節痛を訴える場合には、全人工股関節置換術が必要となることがある。
歯突起低形成 上位頸椎不安定症が認められる、もしくは頸髄障害の臨床所見が認められる場合、上位頸髄の安定化や頸髄圧迫の緩和のために、後頭頸部もしくは上頸部の圧迫解除および癒合手術が必要である。
- 神経損傷を最小化し機能を最大化するために、小児で頸部圧迫のX線所見を認める場合には、症状がなくても治療が推奨される。
- 癒合手術を施行した患者は典型的には回復する。マイナーな二次性合併症としてピン刺入部感染、褥瘡、長期の気管内挿管困難などをきたしうる。
注:臨床医は、上位頸椎不安定症による頸髄障害のため、耐容能の低下や歩行の障害をきたす可能性があると気付くことが重要である。脊髄症が疑われた場合、頸椎の単純X線やMRIを撮影する(「定期検査」の項を参照)。患者を三次医療施設の小児整形外科医/神経外科医に紹介するべきである。
腰椎アライメント不整 (椎体低形成による)胸腰椎後弯症は進行性かつ症候性である可能性がある。
- 45度未満の後弯を認める場合、変形が進行するリスクは弯曲が45度以上の場合よりも小さいが、診察やX線によるモニタリングが必要である。
- 後弯が45度を超える場合。装具/ギプスによる広範囲の支持は胸腰椎後弯症の進行を予防しないが、成長発達がみられる間の外科手術の必要性を遅らせる可能性がある。
- 下記を1つ以上認める場合、前方/後方全周性脊柱固定術(anterior and posterior circumferential spinal fusion)が適応となる。
- 70度を超える進行性の胸腰椎後弯症
- コントロールできない背部痛
- 脊柱管狭窄症に関連する神経学的変化
循環器
心拍数の増加は、小さな左室容積、少ない1回心拍出量を代償する機序である。そのため、β遮断薬による頻脈の治療は避けるべきである。進行性の弁疾患に対し弁置換術を考慮することがある。弁置換術では、人工弁(抗凝固薬を生涯にわたり使用する)、生体弁(弁形成異常・劣化・石灰化のリスクがある)のいずれにするか、リスクを慎重に検討する必要がある。
呼吸器
上気道閉塞や閉塞性睡眠時無呼吸に対しては、肥大した扁桃やアデノイドの切除(平均7歳)を施行する。注:この治療を行っても、なおムコ多糖症の小児患者における閉塞性睡眠時無呼吸の発生率は一般集団よりも高い。それゆえ、迅速に臨床的に評価し、ポリソムノグラフィーを施行することがのぞましい。アデノイド・扁桃切除による上気道閉塞の軽快が一時的でびまん性の気道狭窄を認める患者では、CPAP(持続陽圧呼吸療法)、BiPAP(二相性陽圧呼吸)、気管切開のような他の治療法を考慮する。
下気道閉塞では喘鳴や反復性感染がみられ、気管支拡張薬の吸入や内服、一部はステロイドにより治療を行う。
拘束性肺疾患に対しては支持療法を行う。
成長
ムコ多糖症ⅣA型患児の身長は専用の成長曲線に記入するのが最もよい。
栄養面については、バランスのとれた食事と骨の成長のために十分なビタミンDやカルシウムの摂取を心がけるべきである。
学習環境
身体的な制限があるにもかかわらず、ムコ多糖症ⅣA型患者の知能は正常で、学習および社会的な刺激のある環境では十分な発育が望める。身体的外傷を防ぐための補助があれば、通常は普通学級/学校に通う。
眼
角膜混濁によりしばしば小児期早期に弱視が認められ、全層角膜移植が必要となるが、その成績はさまざまである。移植後1年以内の混濁の再発が報告されており、この場合QOLは一時的にしか改善しない。くわえて、緑内障や網膜症のような他の眼科的な問題により、角膜移植の成功率は低下する可能性がある。白内障を認める患者では、それに対する手術は有用である可能性がある。
歯
毎日の口腔ケア、歯科シーラント、十分なフッ素塗布が齲歯の予防に有用である。不整咬合を治療する歯科矯正が必要となることがある。
聴覚
換気チューブの留置により、慢性的な滲出性中耳炎や反復する急性中耳炎に合併する長期的な瘢痕化のリスクを最小限にし、長期的に聴力を改善することができるため、ほとんどの小児は就学前に換気チューブを留置している。ムコ多糖症ⅣA患者では、滲出性中耳炎の反復や鎮静に伴うリスクがあるため、はじめは長期にわたる鼓膜切開が推奨される。
ムコ多糖腫緒ⅣA型患者のほとんどに進行性の聴力障害が認められ、補聴器が有用である。
一次症状の予防
酵素補充療法に関する情報は「病変に対する治療」を参照。
造血幹細胞移植の経験は極めて限られており、あまり研究されていない。
二次合併症の予防
麻酔 麻酔下に手技を行う場合、念入りな計画が必要である。また、解剖学的異常やグリコサミノグリカンの蓄積、不安定な頸椎、進行性の肺疾患(拘束性および閉塞性)など、ムコ多糖症ⅣA型の気道管理上の問題を経験している麻酔科医が勤務する施設で行うのがもっともよい。モルキオ症候群患児の麻酔に関する最も大規模なコホート研究を行ったTherouxら(2012)は、推奨される麻酔中のケアについて報告している。
術前に、麻酔に対する反応や気道閉塞の兆候などの既往、心電図や心エコーを含む心臓の評価、呼吸機能(スパイロメトリーやポリソムノグラフィー)の評価、気道のX線透視などを行うべきである。
気管内挿管では、ビデオ喉頭鏡、ラリンゲアルマスクエアウェイを用いた/用いないファイバー気管支鏡、年齢や体格に比して小さな挿管チューブが用いられる傾向にある。経鼻挿管も選択肢となるが、グリコサミノグリカンの蓄積により鼻道が狭窄しており、出血が認められる傾向にある。
術後の麻酔は慎重に行うべきである。
睡眠時無呼吸のような、元から存在する呼吸器の問題を悪化させないためには、さまざまな鎮痛剤や麻薬以外の薬剤の使用がのぞましい。
肺水腫のような術後合併症が報告されている。
外科手術 亜急性や活動性の脊柱管狭窄症では脊髄損傷をきたしうるため、頸椎を操作する場合、(脊椎手術を含む)腹臥位で行う場合、(45分を超えるなど)長時間の麻酔を要する場合では、術中神経生理学的モニタリング(intraoperative neurophysiologic monitoring, IONM)を考慮するべきである。術中神経生理学的モニタリングでは、脊髄機能のモニターに体性感覚誘発電位や運動誘発電位を用いる。注:少数の骨系統疾患患者で手術中の脊髄梗塞が報告されているが、このモニタリング技術による改善効果を示すデータは限られている。
予防接種 呼吸器感染症のリスクがあるため、すべての患者は定期接種と同様にインフルエンザや肺炎球菌の予防接種を受けるべきである。
感染性心内膜炎の予防は、人工弁置換術を受けた患者、心臓弁修復術で人工物を使用した患者、感染性心内膜炎の既往があるなどリスクの高い患者において推奨される。
定期検査
酵素補充療法の効果を確かめるため、開始の前後に以下を評価すべきである。
- 少なくとも6ヶ月ごとの身体診察および神経学的評価
- 年1回のQOL、疾病負荷、運動耐容能の評価
- 年1回の最大換気量や努力性肺活量を含む呼吸機能検査
注:(1)尿中ケラタン硫酸値の変化は治療の効果と相関しない。よって、エロスルファーゼアルファ治療中の尿中ケラタン硫酸測定によるフォローの有用性は限られている。(2)抗エロスルファーゼアルファ抗体のモニタリングの有用性は不明である。
すべての患者
QOL、疾病負荷、運動耐容能評価
- 6~12ヶ月ごと:成長・性成熟・発達のフォロー、歩行の最適化。
- 6ヶ月ごとの疼痛評価、1年ごとの年齢別QOL質問票。
- 1年に1回、手術の前後もしくは臨床的に適応があれば、心血管系・呼吸器系・筋骨格系・神経系の機能評価のため6分間歩行検査や25フィート歩行時間測定をふくむ耐容能検査。呼吸数、パルスオキシメーター、心拍数は1年に1回の検査前後に測定すべきである。
筋骨格系 Whiteら(2014)は、ムコ多糖症ⅣA患者の筋骨格系病変のモニタリングについて以下のようなガイドラインを策定した(全文)。
- 上下肢 少なくとも年1回上下肢病変の重症度や進行度を評価する。
- 可動域、握力、ピンチ力の評価や上肢の機能評価(functional dexterity testなど)。
- 以下を含む下肢アライメントの評価:(臨床的に適応がある場合)立位前後像。股関節形成不全/亜脱臼の評価のため、骨が成熟するまで、もしくは臨床的に適応がある場合に骨盤の前後像および蛙肢位側面像。
- 脊椎 脊髄圧迫を評価するため6ヶ月ごとに神経学的評価を行う。
- 小児本人から信頼できる情報を得ることができる場合、受診ごとに運動耐容能や脊髄障害の症状(四肢の筋力低下、不器用さ、不安定で変容した歩行、腸管/膀胱機能障害、腰部/下肢痛など)に関する情報を聴取する。
- 多分節性の脊髄障害を認める患者では、(可能であれば)体性感覚誘発電位や運動誘発電位により詳細な情報を得られる場合がある。
- 6ヶ月ごとに頸椎単純X線(前後像、側面像、中立位、屈曲・伸展位)を行う。
- 1-3年ごとに脊椎単純X線(胸腰椎の前後像・側面像)を行う。
- 全脊椎MRI(中立位)*を年1回、屈曲・伸展位の頸椎MRIを1-3年ごとに行う。
*環椎後頭関節不安定症の評価で、頸椎MRIの前に頸椎X線で中立位・屈曲位・伸展位の側面像を撮影する。
- 小児本人から信頼できる情報を得ることができる場合、受診ごとに運動耐容能や脊髄障害の症状(四肢の筋力低下、不器用さ、不安定で変容した歩行、腸管/膀胱機能障害、腰部/下肢痛など)に関する情報を聴取する。
循環器系 心拍数は毎年評価する。疾患の経過に応じて1~3年ごとに心電図や心エコーを施行する。
呼吸器系
- 閉塞性睡眠時無呼吸に対しては、睡眠のパターンや音に焦点をあてた病歴を聴取する。耳鼻科でアデノイド肥大/扁桃肥大を評価する。在宅でスクリーニングの睡眠検査(酸素飽和度のモニター)を年1回施行する。3年ごとにポリソムノグラフィーを施行する。
- 呼吸機能の評価のため、小児の成長が終了するまで2-3年ごとに最大換気量や努力性肺活量を測定する。ムコ多糖症ⅣA型患児において、非侵襲性の肺機能検査、インパルス・オシロメトリー、胸腹部の動作解析の有用性が示されている。
成長 ムコ多糖症ⅣA型患者用の成長曲線を用いて栄養状態を評価する。受診ごとに身長や体重を測定すべきである。
眼
- 受診ごとに視力や眼の異常についてモニターする。必要に応じて眼科に紹介する。
- 杆体・錐体の網膜変性を認める患者では、5年ごとに暗所視/明所視の網膜検査や網膜電図を行う。
歯
6-12ヶ月ごとに評価する。
聴力
年1回聴力検査(オージオグラム)を施行する。
避けるべき薬物/環境
過剰な体重増加は軸骨格に過度のストレスを及ぼし独立歩行の期間を減らす可能性があるため、生活習慣の改善とともに、成長に適した栄養摂取を行うことが重要である。
心室容積が小さく心拍出量が少ないため、頻脈の治療にβ遮断薬は用いるべきではない。
リスクのある親族の検査
できるだけ早期に発見することで酵素補充療法(「病変に対する治療」を参照)の恩恵を受けることができるため、一見無症状であっても年下の同胞を評価することがのぞましい。以下で評価することができる。
- 家系内の病原性変異が判明している場合には分子遺伝学的検査。
- 家系内の病原性変異が判明していない場合にはGALNS酵素活性の測定。
遺伝カウンセリング目的のリスクのある親族に対する検査に関する問題については「遺伝カウンセリング」の項を参照のこと。
研究中の治療法
軽症患者に対する酵素補充療法(エロスルファーゼアルファもしくはビミジム®)についてはまだ研究中である。参加者は全員機能が高度に保たれている(平均のベースライン歩行距離が非罹患者の70-80%と定義される)MOR-008第Ⅱ相試験で報告されているように、酵素補充療法で運動耐容能や呼吸機能検査にほとんど変化はみられなかったが、運動能力や筋力、疼痛に関していくらか良好な結果が得られている。参加者の18.7%に過敏反応がみられた。全体的に、エロスルファーゼアルファは安全に使用でき重篤な副反応はまれであるが、安全に関するさらに長期的なデータが集められている。
さまざまな疾患に関する臨床試験に関する情報はClinicalTrials.govを参照のこと。
遺伝カウンセリング
「遺伝カウンセリングは個人や家族に対して遺伝性疾患の本質,遺伝,健康上の影響などの情報を提供し,彼らが医療上あるいは個人的な決断を下すのを援助するプロセスである.以下の項目では遺伝的なリスク評価や家族の遺伝学的状況を明らかにするための家族歴の評価,遺伝学的検査について論じる.この項は個々の当事者が直面しうる個人的あるいは文化的、倫理的な問題に言及しようと意図するものではないし,遺伝専門家へのコンサルトの代用となるものでもない.」
遺伝形式
ムコ多糖症ⅣA型は常染色体劣性遺伝性疾患である.
患者家族のリスク
発端者の両親
- 患児の両親は必然的にヘテロ接合体を保有している(すなわち1つのGALNS病原性変異のキャリア)。
- ヘテロ接合体保有者(キャリア)は無症状で、疾患の発症リスクはない。
発端者の同胞
- 受胎時に、罹患者の同胞が罹患している確率は25%、無症候性キャリアである確率は50%、罹患もしておらずキャリアでもない確率は25%である。
- ヘテロ接合体保有者(キャリア)は無症状で、疾患の発症リスクはない。
発端者の子ども
ムコ多糖症ⅣA型患者の子どもは必然的にGALNS病原性変異のヘテロ接合体保有者(キャリア)である。
他の家族
発端者の両親の同胞は、50%の確率でGALNS病原性変異のキャリアである。
保因者診断
リスクのある親族に対する保因者診断を行うには、先に家系内のGALNS病原性変異の同定を行う必要がある。
遺伝カウンセリングに関連した問題
早期診断・治療を目的としたリスクのある親族の検査についての情報は、「臨床的マネジメント」「リスクのある親族の検査」を参照のこと。
家族計画
- 遺伝学的リスクの評価や保因者診断を行う、および出生前診断の利用について話し合いをするのに最適な時期は妊娠前である。
- 罹患している、キャリアである、もしくはキャリアであるリスクのある若年成人に対して遺伝カウンセリング(潜在的な子どもへのリスクや出産方法の選択肢に関する話し合いなど)を申し出ることがのぞましい。
DNAバンクは(主に白血球から調整した)DNAを将来利用することを想定して保存しておくものである。検査技術や遺伝子、アレル変異、あるいは疾患に対するわれわれの理解が将来さらに進歩すると考えられるので、患者のDNA保存を考慮すべきである。
出生前診断および着床前診断
家系内での病原性変異が同定されている場合、ムコ多糖症Ⅳ型のリスク妊娠における出生前診断や着床前診断は実施可能な選択肢である。
とくに検査が早期診断ではなく妊娠中絶を目的とした場合に、医療従事者や家族の間で出生前検査に関して視点の違いが存在する可能性がある。出生前診断に関する決定は両親の選択によるが、これらの問題について話し合うことがのぞましい。
更新履歴
- Gene Reviews著者: Debra S Regier, MD, PhD, Matthew Oetgen, MD, and Pranoot Tanpaiboon, MD.
日本語訳者: 和田宏来(県西総合病院小児科/筑波大学大学院小児科)
Gene Reviews 最終更新日: 2016.3.24.日本語訳最終更新日:2017.6.29.(in present)