肥大型心筋症概説
(Hypertrophic Cardiomyopathy Overview)
GeneReviews著者: Allison L Cirino, MS, CGC , Carolyn Ho, MD.
日本語訳者:武井 眞(東京都済生会中央病院循環器内科)
GeneReviews最終更新日: 2021.7.8 日本語訳最終更新日: 2021.7.12
原文 Hypertrophic Cardiomyopathy Overview
要約
本概説の目的は臨床家の肥大型心筋症(Hypertrophic cardiomyopathy, HCM)の遺伝的背景に対する認識を高め、遺伝性HCMの症例に対して早期の診断と介入によるメリットを提供することである。
目標1.
HCMを定義すること
目標2.
HCMの分類を理解すること
目標3.
HCMの発端者に(可能であれば)特定の遺伝学的原因診断を確立するための評価を提供すること
目標4.
HCM発端者の無症状の親族に対して遺伝的リスクの評価を行い、心臓サーベイランスの提供、早期のHCMの診断および治療を可能とすることで長期予後を改善すること
1.肥大型心筋症の定義
肥大型心筋症(hypertrophic cardiomyopathy, HCM)は,通常,成人においては最大左室壁厚15mm以上、小児においてはZ-score 3以上の原因不明の左心室肥大(left ventricular hypertrophy, LVH)によって定義される[Gersh et al 2011, Elliott et al 2014].HCMの家族歴がある場合、もしくは遺伝学的検査により該当親族が家系内の病的サルコメアバリアントを継承していることが確かめられた場合は左室壁厚13mm以上でも本疾患の診断を考慮する。HCMのLVHは, 圧負荷または蓄積性/浸潤性疾患のような,左室壁肥厚を来たしうる循環器疾患あるいは全身性疾患を認めず、左室拡大も認めない.
HCMの診断は通常心エコー検査や心臓の磁気共鳴画像(心臓MRI)を含む非侵襲的な心臓画像処理によって確定される.
- 非対称性心室中隔肥大は,肥大の中で最も一般的なパターンであるが,肥大の程度と位置はさまざまである.LVHはびまん性、求心性であることもあれば心筋局所、心尖部などに限局して見られることもある。
- 経胸壁心エコー検査では以下の所見が見られることがある:
- 左室流出路狭窄や僧帽弁閉鎖不全に関連した僧帽弁の収縮期前方運動(systolic anterior motion, SAM)
- 収縮期心室腔閉塞の結果としての左室体部狭窄
- 拘束型パターンを含む拡張障害.左室拡張障害はサルコメアの構成要素に病的バリアントを有する症例においては左室壁厚が正常であっても認められることがあり[Nagueh et al 2001,Ho et al 2002],拡張障害は,LVHの二次的な結果というよりもHCMの初期表現型であることを示唆している.
LVHやHCMの臨床診断はしばしば思春期開始前後の青年期や若年成人期に明らかになるが、発症時期はより早期(乳児または小児期)であったり、晩期であることもある。[Niimura et al 2002]. 一般的な症状には、息切れ(特に労作時)、胸痛、動悸、起立性低血圧、前失神、失神などがある。
表現型のばらつき及びその進行
HCMの臨床的な表現型には高度のばらつきがあり、無症状のLVHから不整脈(心房細動のみならず致死性の心室性不整脈まで)、治療抵抗性の心不全まで多様である。さらに、同一家系内においても表現型が異なっていることがある。
HCM症例のおよそ1/3は安静時に検知しうる心室内狭窄を有し、別の1/3は誘発(前負荷、後負荷の低減)により流出路狭窄を示すとされる[Maron et al 2003, Elliott et al 2014].流出路狭窄の重症度は必ずしも厳密に症状の重症度や心臓突然死のリスクと相関しているわけではない。一連の観察研究からは流出路狭窄を有するHCM症例では有さない症例に比べて症状の進行、脂肪のリスクが高い可能性があると報告されている[Maron et al 2003, Sorajja et al 2009];。高圧較差は長期間にわたって代償されることもありうる。
HCM症例は心房細動(atrial fibrillation; AF)の発症リスクが高くなる。心房細動は全身性塞栓症、症状の悪化のリスクを高め、予後に大きな影響を及ぼす。心房細動の有病率は年齢とHCMの罹患機関に従って増加する。HCM症例全体での心房細動の有病率は20%以下とされるが、40歳以下で診断されたHCM症例の60%が60歳までに心房細動を発症すると報告されている。[Elliott et al 2014, Ho et al 2018].HCMと心房細動を合併した症例では塞栓性合併症の有病率は27%と見積もられている。[Elliott et al 2014].
約5~10%のHCM患者は,収縮障害の進行とともに進行性の末期状態に移行し,一部の例においては,左室の拡張及びLVHの退縮が見られることがある.末期患者の年間死亡率は11%と推定され[Harris et al 2006],心臓移植が必要とされることがある.
ほとんどの場合は心室頻拍/細動に関連してみられるが、心臓突然死(Sudden Cardiac Death; SCD)は比較的稀ではあるが重要なHCMの転帰である。
- 大規模なコホート研究において、HCM症例の6%が心臓突然死、蘇生された心肺停止、適切なICD作動を経験した[Ho et al 2018].
- SCDは病気の最初の臨床症状となることがある[Maron et al 2000, Finocchiaro et al 2019].
- アメリカ合衆国においては競技スポーツを行うアスリートにとって、HCMはSCDのリスクであることが確立しているが[Maron 2003],最近の報告ではその頻度は低下(5%以下から14%)しているようである。[Eckart et al 2011, Harmon et al 2014, Bagnall et al 2016].
- 突然死は青年または若年成人で最も頻繁に発生するが、どの年齢でも発生する可能性があり、リスクは生涯にわたって持続する。
寿命
米国の一般人口と比較して、HCMのある個人の死亡率は約3倍高いが、20〜29歳のHCMの若年症例の死亡率は4倍も高くなっている。突然死は死亡の16%を占める。[Ho et al 2018].
2.肥大型心筋症の分類
(遺伝性の)HCMの鑑別診断としては後天性のLVH、LVHを表現型として含む先天性症候群(他臓器にも所見を有する)もしくは非症候群性(他臓器に所見を有さない)の先天性LVHがあげられる。
後天性(二次性)左室肥大
二次性LVHは圧負荷(全身性高血圧,大動脈弁狭窄症など)に反応して生じ、病的になりうる.このタイプの病的なリモデリングは拡張障害、心不全を惹起しうる。生理的な肥大(アスリート心)は高度のアスレティックトレーニングの結果として生じうる。こうしたトレーニングによる心臓のリモデリングは左室肥大だけでなく左室拡大を伴ことが多い。こうした生理学的なリモデリングは正常な適応の結果であり、有害事象の発生とは関連しないと考えられている。病的、生理的な二次性LVHはいずれも原因が除去されることにより退縮しうる(高血圧の治療やアスリートにとってのトレーニングの中断期間)
先天性症候群に伴うHCM(他臓器にも所見を有する)
GeneReviewsでは、a)特定の診断名を示唆する(分子遺伝学的検査により確定されうる)もしくはb)確定的分子遺伝学的所見がない状態でも診断を確定できる一連の多臓器の臨床所見を示すものを症候群と定義する。先天性症候群に伴うLVHの一部のリストをTable1.に示す。本概説では先天性症候群に伴うHCMについて以下の表以上の言及は行わない。
Table1. 先天性症候群に伴うHCM
| 疾患名 | 原因遺伝子 | 遺伝形式 | 臨床的特徴 |
|---|---|---|---|
| Danon 病 | LAMP2 | XL | 骨格筋ミオパチー 網膜ジストロフィー |
| Fabry 病 | GLA | XL | 周期的な四肢の痛み 被角血管腫 無汗症 眼科的異常 蛋白尿・腎機能障害 |
| Friedreich 失調症 | FXN | AR | 25歳以前の緩徐進行性失調 構音障害 筋委縮 |
| 先天性致死性糖原病心疾患l(OMIM 261740) | PRKAG2 | AD | 新生児期の低血糖 空胞性ミオパチー 軽度の顔面奇形/巨舌 |
| 遺伝性トランスサイレチンアミロイドーシス | TTR | AD | 緩徐進行性の末梢運動、感覚、自律神経 のニューロパチー 硝子体混濁 中枢神経アミロイドーシス |
| Pompe 病 | GAA | AR | 感覚障害 巨舌 運動発達遅滞/筋委縮 呼吸障害 |
RASopathies 2 (以下を含む)
|
BRAF,HRAS, KRAS,LZTR1, MAP2K1,MAP2K2, NRAS,PTPN11, RAF1,RASA2, RRAS,RIT1, SOS1,SOS2 |
AD | 特徴的顔貌 低身長 種々の発達遅滞 翼状頚 胸郭奇形 |
AD = 常染色体顕性遺伝; AR = 常染色体潜性遺伝; XL = X連鎖
- アルファベット順
- RASopathies とは共通する病的な遺伝的背景によって生じる一連の症候群で、その臨床的表現型は一部重複する。 [Tidyman & Rauen 2009].
先天性HCM(先天性症候群に分類されない)
後天性HCMもしくは先天性症候群に伴うHCM(Table1)は先天性症候群に伴わない先天性HCM(GeneReviewでは他臓器病変を伴わないHCM)に含まれない。以降のこのGeneReviweにおいて、他臓器の病変を伴わないHCMの原因となる遺伝子はHCM遺伝子と称する。現在知られているHCM遺伝子のリストをTable2に示す。HCMとそれぞれの遺伝子の関連性の強さはさまざまである[Ingles et al 2019]。最も臨床的に強くHCMと関連する遺伝子群はサルコメアの異なった構成因子をコードしている[Ingles et al 2019]。
サルコメアを構成するいくつかの遺伝子の病的バリアントは家族歴のあるHCM発端者(成人及び小児を含む)の50-60%、孤発例の20-30%に認められる[Alfares et al 2015]。罹患者の約3-5%は二つ以上のサルコメア遺伝子のバリアントを有している(一つの遺伝子の両アレルにバリアントを有しているものと複数遺伝子にバリアントを有しているもの双方を含んで)が、病的もしくは病的である可能性が高いバリアントを複数有している確率は1%以下とされる[Alfares et al 2015, Burns et al 2017].。
表2. HCM遺伝子
| 遺伝子1 | 遺伝形式 | HCMに占める当該病的バリアントの割合2 | ClinGenによる臨床的妥当性分類 | 同一アレル疾患 3 | 参考文献/OMIM Gene Entry |
|---|---|---|---|---|---|
| MYBPC3 | AD | 50% | Definitive | 拡張型心筋症 | 600958 |
| MYH7 | AD | 33% | Definitive | Laing 遠位型ミオパチー ミオシン蓄積ミオパチー 左室緻密化障害 肩甲骨腓骨筋ミオパチー 拡張型心筋症 |
160760 |
| TNNI3 | AD | 5% | Definitive | 拡張型心筋症 拘束型心筋症 |
191044 |
| TNNT2 | AD | 4% | Definitive | 拡張型心筋症 左室緻密化障害 家族性拘束型心筋症 |
191045 |
| ACTC1 | AD | <3% | Definitive | 拡張型心筋症 | 102540 |
| MYL2 | AD | <3% | Definitive | 拡張型心筋症 | 160781 |
| MYL3 | AD/AR | <3% | Definitive | 160790 | |
| PLN | AD | <3% | Definitive4 | 拡張型心筋症 不整脈原性右室心筋症 |
172405 |
| TPM1 | AD | <3% | Definitive | 拡張型心筋症 | 191010 |
| ALPK3 | AR | 稀 | Strong | 617608 | |
| ACTN2 | AD | <1% | Moderate4 | 拡張型心筋症 | 102573 |
| CSRP3 | AD | <1% | Moderate | 拡張型心筋症 | 600824 |
| TNNC1 | AD | <1% | Moderate | 拡張型心筋症 | 191040 |
| JPH2 | AD | 稀 | Moderate | 拡張型心筋症 | 605267 |
| MYOZ2 | AD | <1% | Limited | 605602 | |
| NEXN | AD | <1% | Limited | 拡張型心筋症 | 613121 |
| ANKRD1 | AD | 稀 | Limited | 拡張型心筋症 | |
| CALR3 | AD | 稀 | Limited | 611414 | |
| KLF10 | AD | 稀 | Limited | 601878 | |
| MYH6 | AD | 稀 | Limited | 拡張型心筋症 | 160710 |
| MYLK2 | 2遺伝子遺伝 | 稀 | Limited | 606566 | |
| MYOM1 | AD | 稀 | Limited | 603508 | |
| MYPN | AD | 稀 | Limited | 拡張型心筋症 ネマリンミオパチー |
608517 |
| PDLIM3 | AD | 稀 | Limited | 605889 | |
| RYR2 | AD | 稀 | Limited | カテコラミン誘発性多型性心室頻拍1型 | 180902 |
| TCAP | AD | 稀 | Limited | 拡張型心筋症 肢帯筋ジストロフィー2G |
604488 |
| TRIM63 | AD | 稀 | Limited | 606131 | |
| TTN | AD | 稀 | Limited | 拡張型心筋症 早発性呼吸不全を伴う遺伝性ミオパチー 肢帯筋ジストロフィー2J Salih ミオパチー Udd 遠位型ミオパチー |
613765 |
| VCL | AD | 稀 | Limited | 拡張型心筋症 | 193065 |
AD = 常染色体顕性遺伝; AR = 常染色体潜性遺伝
- 表の順序は臨床的妥当性分類、HCMの原因としての頻度、アルファベット順に従った。
- これらの遺伝子の頻度は Alfares et al [2015]を参照. "稀" と記載したものはこの論文に記載が認められなかったものである。
- 同一遺伝子の病的バリアントによって引き起こされる他の表現型
- PLN と ACTN2 は孤発性LVHとHCMを含む多様な心臓表現型との関連が示されていることから、原発性心筋症についてこのように分類されている。
3.(可能な場合における)肥大型心筋症の特定の遺伝的背景の確立
遺伝学的検査は①HCMの診断基準を満たす罹患者において血縁者のスクリーニングを可能とするため②HCMの診断が疑われる症例における確定診断のため、の場合に推奨される。[Elliott et al 2014].
HCMの分子遺伝学的な診断を確立する目的としては、①診断により治療方針が変更される可能性のある先天性症候群に伴うHCMの同定②発端者の親族にリスク評価について情報提供をする(遺伝リスクの評価及び心血管系サーベイランスを参照)。評価の対象となる個人の病歴及び家族歴の一部(HCMの家族歴、中隔の形態、若年での診断)は遺伝学的検査が陽性となる確率と関連している。
肥大型心筋症(HCM)を持つ個人の特定の遺伝的要因を識別する一般的なアプローチは,図1の通りである.HCMに対する遺伝学的検査は個人よりは家系を対象とするものとして捉えるのが適切である。なぜならば、遺伝学的検査の結果をより適切に解釈するためには複数の家系内の被検者に対して遺伝学的検査の結果を臨床的な検査(心電図や心臓超音波検査)と統合することが必要だからである。こうした方法をとることにより、家系内の臨床的表現型の包括的な理解をバリアントが表現型とともに分離されているか確認することに利用できる(病的であると疑われるバリアントは罹患している家系内の被験者には認められ、そうでない被験者には認められてはならない)
初期の評価は以下を含むべきである。
家族歴:
詳細な3,4世代の家族歴を入手する.親族において次のいずれかの既往歴の有無に特に注意するべきである:心不全,HCM, 心臓移植,原因不明の死亡または突然死(特に40歳前の血縁者),心臓伝導系疾患や不整脈,原因不明の脳卒中または他の血栓塞栓性疾患.注:分子遺伝学的検査を受ける家族の一員は,HCMの確定診断を必要とする,理想的には家系内で最も明確に表現型を示す個人である.
注:(1)様々な医学的問題(例えば適切な心臓スクリーニングを受けていない;低浸透率;HCM発症前の他の原因による早期死亡)あるいは社会的要因(例えば家族からの孤立,非公開の養子縁組,代替父親または母親)によって親族が本当に一人もHCMに罹患していないか確認することが困難である場合がある。したがって,HCMの発端者が本当に孤発性(すなわち家族内でひとりだけの発症)かどうかを区別できない場合がある.(2)家系構成員の臨床的評価は,家族歴についての貴重な情報を提供できる(たとえばこれまでHCMと認識されていなかった家系構成員の診断).(3)家族歴は見直され,定期的に更新されるべきである.
遺伝カウンセリング は遺伝学的検査が行われるかどうかにかかわらずHCMのすべての症例に対して推奨される。[Elliott et al 2014].
HCMの分子遺伝学的検査は包括的な(HCMに関連することが知られている遺伝子に加え多様な遺伝性心筋症に関連する遺伝子も含む)マルチジーンパネルに依存している。現時点でのHCMと関連することが知られている遺伝子のリストは表2を参照。
表2に示されている臨床的妥当性が確立された遺伝子を含むHCMマルチジーンパネルを用いることが最も妥当なコストで、臨床的意義の不明確なバリアントや臨床的表現型を説明できない遺伝子における病的バリアントの検出を最小限に抑えつつ病態の遺伝的な原因を同定できる可能性が高い方法である。注:1)検査機関によってパネルに含まれる遺伝子の種類、検査精度が異なる可能性がある。また、これらの事柄は経時的にも変化すると考えられる。2)いくつかのマルチジーンパネルにはGeneReviewで取り上げている病態と関連しない遺伝子が含まれている可能性がある。3)いくつかの検査機関ではパネルオプションとして検査機関によってデザインされたパネルや臨床家によって指定された遺伝子を含む表現型に注目したエクソーム解析を提供している。4)パネル検査で行われる解析手法にはシークエンス、欠失/重複解析、シークエンスを用いない遺伝子解析が含まれる。
HCMの検査においては重複/欠失解析を含むマルチジーンパネルが推奨される(表2を参照)
マルチジーンパネル についての基本的な情報はこちらを、遺伝学的検査を依頼する臨床家に向けた詳しい情報はこちらを参照。
HCMのマルチジーンパネル解析で確定的な結果が得られない場合、エクソーム(もしくはゲノム)シークエンスも考慮可能であるが、期待される上積み効果は低い。[Cirino et al 2017b].もしエクソームシークエンスが診断的でない場合、(臨床的に使用可能であれば)エクソンシークエンスでは検出不可能な(複数の)エクソン欠失や重複を検出できるエクソームアレイも検討可能である。
包括的な遺伝学的検査についての基本的な情報はこちらを、遺伝学的検査を依頼する臨床家に向けた詳しい情報はこちらを参照。
遺伝学的検査を依頼する医療従事者はHCMの遺伝学に精通していることが求められる。遺伝学的検査の結果解釈は複雑で、患者のサーベイランスや臨床的マネジメントに及ぼす影響を考慮すると心血管遺伝センターや心血管疾患の遺伝学に精通した遺伝カウンセラーへの紹介を検討する必要がある。(MSGC-Find a Genetic CounselorやABGC Find a Certified Genetic Counselorなどで検索可能)それぞれのバリアントの臨床的意義づけのカテゴリーに対する基本的なガイダンスは表3を参照すること。
表3
| バリアント分類 | |||||
|---|---|---|---|---|---|
| 良性もしくはバリアントなし | 良性の可能性が高い | 臨床的意義不明 | 病原性の可能性が高い | 病原性 | |
| 意義 | 陰性 | 不明 | 陽性 | ||
| 臨床的に重要なバリアントは検出されない。遺伝性疾患の除外はできない | 検知されたバリアントは無害である可能性が高い。遺伝性疾患は除外できない | 未確定な結果。当該バリアントの病原性についてのデータが乏しい。表現型と当該バリアントの家系内での分離を評価することにより追加情報が得られる可能性がある。 | HCMの原因となっている可能性が高い。表現型と当該バリアントの家系内での分離を評価することにより追加情報が得られる可能性がある。 | HCMの原因となっている | |
| 発端者への有用性 | なし | なし | 不明 | HCMの診断を示唆する。臨床的マネジメントの変更、追加検査につながる可能性がある。 | HCMの確定診断。臨床的マネジメントに追加情報を与える可能性がある。 |
| 血縁者への有用性 | 発症前診断は施行困難。 経時的な臨床表現型の評価に依存する。 |
発症前診断は施行困難。 経時的な臨床表現型の評価に依存する。 |
発症前診断は推奨されない。 経時的な臨床表現型の評価に依存する。 表現型を示す血縁者に対して遺伝型との分離を確認することを検討する。 |
発症前診断は注意深く検討し、臨床表現型の評価及び定期的なサーベイランスとともに提供することを検討する。 | 発症前診断に使用可能 |
Cirino et al [2017a]より引用
4.リスク親族において早期治療可能なHCMの表現型を検知するための遺伝的リスク評価
ガイドラインではリスク親族を同定するために少なくとも三世代の家系図をHCMのすべての症例について作成すべきであると推奨している。[Hershberger et al 2018].リスク親族は表4にあげられているガイドラインに沿った臨床的評価を受けるべきであり、家系内で病原性バリアントが同定されていれば遺伝学的検査も考慮することができる。未発症の小児に対する検査の時期は潜在的な利益と害を比較したうえで慎重に検討されるべきである。
もし家系内で同定されたバリアントの病原性が不確かならば(すなわち,病原性らしいまたは臨床的意義不明),家系内解析を行うことが変異の意義の解釈の手助けとなりうる。すなわち、HCMに罹患している他の家族が同定されたバリアントを有していれば(そして健常者は有していなければ),さらに病原性の裏付けとなる.
第一度近親者がバリアントを受け継ぐ可能性は50%であるので検査した血縁者の数は重要な検討事項である.一方,罹患しているがそのバリアントを持たない者がひとりでもいれば,このバリアントが病原性ではないことの強い証拠となる.
・疾患と家系内のバリアントの伝わり方について,より包括的な情報を提供するために,リスクのある家系の臨床的な評価と遺伝学的検査を組み合わせることは適切である.
・あるバリアントの病原性が,家系内解析により否定された時には,遺伝学的検査施設にこの情報を伝えるべきである.
検査をうけた家族に同定されたバリアントが臨床的意義不明であるならば,罹患していない血縁者の検査は役に立たない.こうした情報はバリアントの解釈の助けにはならず,血縁者がHCMを発症する事前リスクが変更されることもないからである.
検査を受けた家族で何らバリアントが特定されないならば,リスクのある家系の遺伝学的状況を明らかにするためのさらなる遺伝学的検査は(その時点では)行いえない。
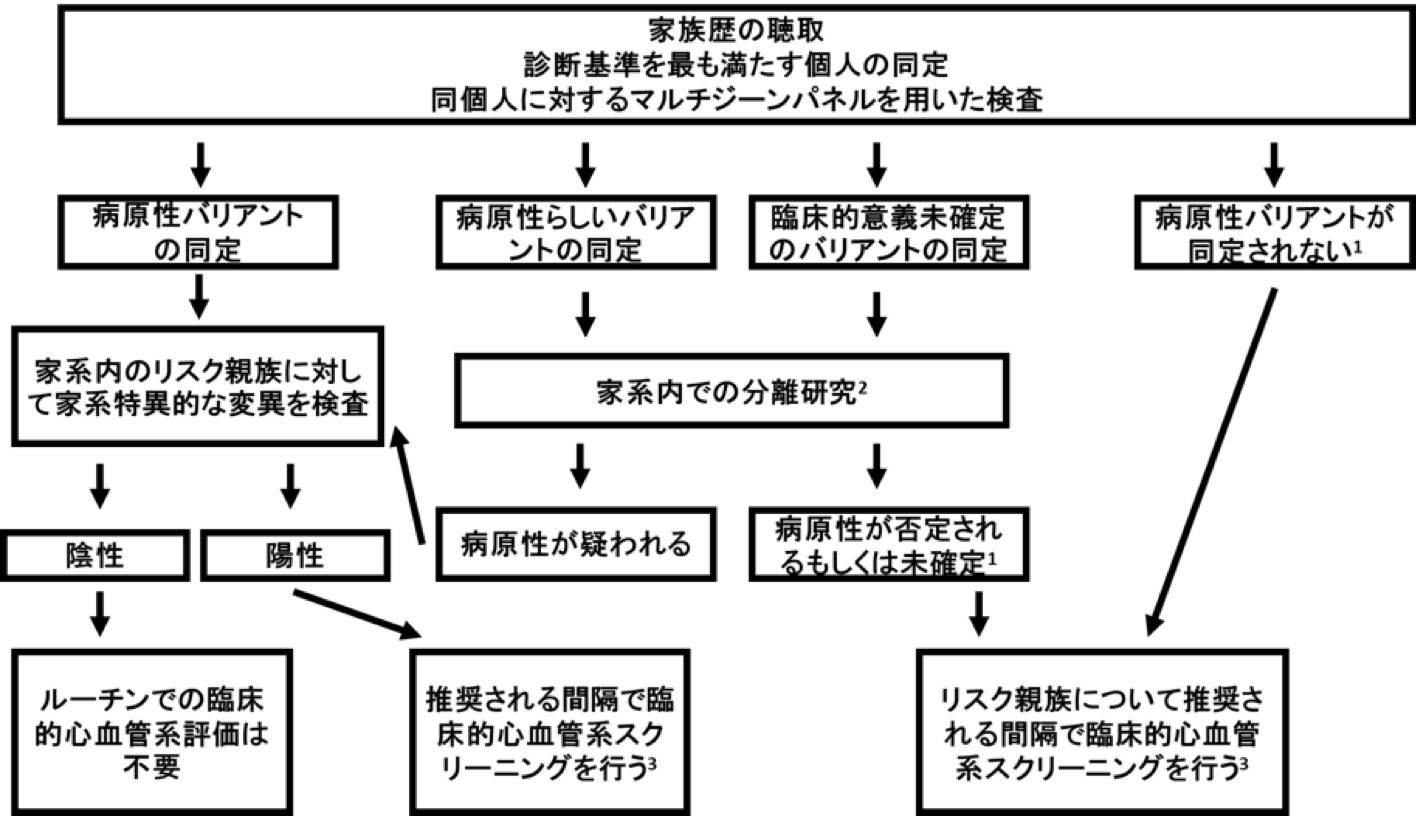
図1:家族性HCM: 遺伝学的検査及び臨床的心血管スクリーニングのアルゴリズム
注:
- 重要バリアントは検出されないが遺伝性疾患は除外できない。新しい遺伝子/検査が利用できる時,診断基準を満たす患者の再検査を検討する
- 家系内におけるバリアントと表現型の分離についてフィードバックを遺伝子検査機関に提供する
- 表4.健康なリスク親族の臨床スクリーニングのためのガイドラインを参照。(注:病原性バリアントが確認された罹患者の血縁者と確認されなかった罹患者の無症候性の第一度近親者(成人と子ども)両者にこれらのガイドラインが適応される)
遺伝リスクの評価
HCMは典型的には常染色体顕性遺伝の形式をとる。常染色体潜性遺伝を呈する遺伝子の病的バリアントも報告されているが、稀である。
稀ではあるが同一症例においてサルコメアタンパクを構成する一つの遺伝子に複数の病的バリアントが同定されることがある(二重ヘテロ接合)[Alfares et al 2015, Burns et al 2017]。したがって、一症例についてそれぞれのバリアントの遺伝形式を決定することが家系内での正確なリスク評価に重要である。
家系内でのリスク
常染色体顕性遺伝
発端者の両親
- HCMであると診断された一部の患者は罹患した親を持つ.
- 発端者は新生突然変異の結果として,HCMに罹患している可能性がある。新生突然変異による罹患者の割合は明らかではない。
- 明らかに新生突然変異によると思われる発端者の両親に対する推奨される評価法には,発端者で同定されている病的バリアントに対する遺伝学的検査、心エコー,心電図,HCMに精通した心臓専門医による検診が含まれる.
- 発端者において同定された病的バリアントが両親いずれの白血球DNAからも検出されない場合、その病的バリアントは発端者において生起した新生突然変異の可能性が高いが、両親のいずれかが性腺モザイクを有している場合、発端者はその親から病的バリアントを受け継いだ可能性が残る。性腺モザイクの症例が一例報告されているが、正確な発生率は不明である。[Forissier et al 2000]
注:一部の家族性HCMではHCMに罹患した親が実際には存在するにもかかわらず,家族が無症状あるいは軽微な症状,親の発症前の早期死亡,遅い発症などによって疾患の確認ができず,家族歴が陰性に見える場合がある。したがって、適切な臨床的評価および、もしくはそれに追加して分子遺伝学的評価が発端者の両親に行われるまでは家族例が陰性と確定することはできない。
発端者の同胞
同胞のリスクは発端者の両親の遺伝的状態による.
- 発端者のどちらかの親が病的バリアントを有していた場合、同胞が病的アレルを継承するリスクは50%である.しかしながら臨床的重症度と発症年齢を予測することは困難である。
- もし発端者で認められた病原性変異がどちらの親の白血球DNAにも検出されない場合、理論的な両親の性腺モザイクの可能性を含めて同胞のリスクは1%以下と推定される[Rahbari et al 2016]。
発端者の子
家族性HCM罹患者の子は50%の確率で病的バリアントを受け継ぎ,HCMを発症するリスクがある.しかしながら浸透率は不完全である場合があり,疾患の重症度や発症年齢は予測できない.
他の家族
他の家族のリスクは,発端者の両親の状態による.もし親が罹患もしくはHCMとの関連が報告されている病的バリアントを有する場合,その親の血縁者にはリスクがある.
常染色体潜性遺伝
発端者の両親
- ある罹患者の両親は定義上ヘテロ接合(HCMの原因遺伝子の病的バリアントの保因者)である。
- 典型的には保因者の発症リスクは一般人口と同様である。しかしながら、ALPK3の保因者は成人期に心筋症を発症するリスクがあるかもしれない[Almomani et al 2016]。
発端者の同胞
- 概念的にはそれぞれの同胞が罹患者となる確率が25%、保因者となる確率が50%、病的バリアントを有さない可能性が25%である。
- 典型的には保因者の発症リスクは一般人口と同様である。しかしながら、ALPK3の保因者は成人期に心筋症を発症留守リスクがあるかもしれない[Almomani et al 2016]。
発端者の子供
常染色体潜性遺伝のHCM発端者の子供は定義的に該当バリアントの保因者となる。
他の家族
発端者の両親の同胞はそれぞれ該当病的バリアントの保因者である可能性が50%となる。
心血管系サーベイランス
HCM発症リスクを有する家系内の個人に対して疾患の発症前に臨床的、遺伝的評価を行い、無症候性のHCM診断、未発症者の早期診断を可能とすることは適切である。
HCMと関連することが知られている病的バリアントを有する罹患者の血縁者
確定的な病的バリアントが罹患者において同定されている場合、リスク親族の遺伝学的状態を明らかにするために発症前遺伝学的検査を行うことができる。病的バリアントのヘテロ接合であることが確認された家系内の個人(HCMを発症する可能性が高い)に対しては公表されている推奨に則り身体所見、心電図、心臓超音波検査を含む臨床的な心血管系評価を行う[Ommen et al 2020](表4aを参照)
表4a: HCMと関連することが知られている病的バリアントを有する罹患者の血縁者に対する臨床的スクリーニングのガイドライン
| 遺伝状態 | 無症候血縁者の年齢 1 | HCM発症リスク | スクリーニング開始時期 | 心電図、心臓超音波検査の間隔2 |
|---|---|---|---|---|
| HCM関連病的バリアントのヘテロ接合 | 小児/若年成人 | 高3 | 家系内でHCMと診断された罹患者が確認された時点 | 1-2年ごと |
| 成人 | 3-5年ごと | |||
| HCM関連病的バリアントのヘテロ接合でない | 小児/若年成人 | リスク上昇なし | 心血管系スクリーニングは必須ではない | データなし |
| 成人 |
Ommen et al 2020から引用
- 第一度近親者を指す。臨床兆候によってはそれより血縁として遠い親族も含む。
- 家族歴、臨床兆候を参照に調整する。
- HCM関連病的バリアントのヘテロ接合であるリスク親族は15年のフォローアップで50%の浸透率を示したと報告されている。[Lorenzini et al 2020]
HCMの特異的な原因遺伝子が特定されていない罹患者の血縁者
病的バリアントが特定されていないHCM罹患者の全ての無症候性の第一度近親者(小児と成人を含む)に公表されている推奨に則り身体所見、心電図、心臓超音波検査を含む臨床的な心血管系評価を行う[Ommen et al 2020]
表4b: HCMの特異的な原因遺伝子が特定されていない罹患者の無症候性血縁者に対する心血管系スクリーニングのガイドライン
| 無症候血縁者の年齢 1 | 家系内の罹患者の発症年齢 | スクリーニング開始時期 | 心電図、心臓超音波検査の間隔2 |
|---|---|---|---|
| 小児及び若年成人 | 幼児期、小児期の発症 | 家系内でHCMと診断された罹患者が確認された時点 | 1-2年ごと |
| 思春期中もしくは以降の発症 | 家系内でHCMと診断された罹患者が確認された時点、遅くとも思春期までに開始 | 2-3年ごと | |
| 成人 | 年齢を問わない | 家系内でHCMと診断された罹患者が確認された時点 | 3-5年ごと |
Ommen et al 2020から引用
- 第一度近親者を指す。臨床兆候によってはそれより血縁として遠い親族も含む。
- 家族歴、臨床兆候を参照に調整する。
診断に役立つような臨床所見(左室肥大など)の浸透率は年齢に依存するため、一度の評価でHCMの将来の発症リスクを除外することはできない。診断的な臨床的表現型は幼児期や小児期には見られないことが多く、若年成人もしくはそれ以降に明らかになってくることが多い。したがって、家族歴、対象者の年齢、臨床医の判断に基づいた頻度で長期的なフォローアップが必要とされる。新たな臨床的病態や兆候が生じた際にはその時点でスクリーニングを行うべきである。
注:ヒトDNAの多様性についての理解が進展することにより、それぞれのバリアントに対する臨床的意義づけが変化し、それらを有する個人や血縁者に対する推奨が変化することが起こりうる。遺伝学的検査の手法も進歩しているため、HCM罹患者の血縁者のフォローアップは定期的に循環器遺伝外来もしくは遺伝カウンセラーとともに行うべきである
参考情報
- Children's Cardiomyopathy Foundation (CCF)
PO Box 547
TenAFly NJ 07670
Phone: 866-808-2873 (toll-free)
Fax: 201-227-7016
Email: info@childrenscardiomyopathy.org
www.childrenscardiomyopathy.org
- Hypertrophic Cardiomyopathy Association (HCMA)
328 Green Pond Road
PO Box 306
Hibernia NJ 07842
Phone: 973-983-7429
Fax: 973-983-7870
Email: support@4HCM.org
www.4HCM.org
- My46 Trait Profile
Hypertrophic cardiomyopathy
・American Heart Association (AHA)
7272 Greenville Avenue
Dallas TX 75231
Phone: 800-242-8721 (toll-free)
Email: review.personal.info@heart.org
Hypertrophic cardiomyopathy
- Cardiomyopathy UK
Chiltern Court
Asheridge Road
Unit 10
Chesham Buckinghamshire HP5 2PX
United Kingdom
Phone: 0800 018 1024 (UK only); 0800 018 1024 (UK only)
Email: info@cardiomyopathy.org
www.cardiomyopathy.org
- Sarcomeric Human Cardiomyopathy Registry (SHaRe)
Email: share@myokardia.com
www.theshareregistry.org
更新履歴:
- Gene Review著者: Allison L Cirino, MS, CGC , Carolyn Ho, MD.
日本語訳者: 宮﨑 幸子(札幌医科大学大学院医学研究科修士課程遺伝カウンセリングコース),櫻井晃洋(札幌医科大学医学部遺伝医学)
GeneReviews最終更新日: 2014.1.16. 日本語訳最終更新日: 2016.11.14. - GeneReviews著者: Allison L Cirino, MS, CGC , Carolyn Ho, MD.
日本語訳者:武井 眞(東京都済生会中央病院循環器内科)
GeneReviews最終更新日: 2021.7.8 日本語訳最終更新日: 2021.7.12[in present]
![]()